


























以下の文章は医薬品医療機器レギュラトリーサイエンスPMDRS誌51巻1号22-26頁に掲載された総説をカラー化したものです。PDF版はここからダウンロードできます。
はじめに
製薬企業の研究員と話をするたびに「創薬標的が枯渇している」と聞く。近年、ヒト病態に関与する遺伝子の特定から、当該遺伝子の欠失による機能評価や遺伝子産物に結合するリガンドの探索によって治療標的が次々とスクリーニングされ、そうしたゲノム創薬が抗体医薬や分子標的薬の急増に見られるような新医薬品の多様化を実現できた当然の成り行きである、と私は思っている。しかし現実に、低分子化合物を設計する従来型の創薬は困難に直面している。この状態を打開するために生み出されたアイデアの1つがドラッグリポジショニング(DR)である。
承認薬の有効成分は後述するように今や5000種類以上も存在するが、前臨床であらゆる薬理作用点への結合活性を確認しているわけではない。これは化合物の遺伝子発現への影響がRNA sequencingなど1実験で網羅的に得られるのに比べて、各作用点に対する結合活性はハイスループットスクリーニング(HTS)を個別に組み立てる必要があり、高コストかつ多大な労力を必要とするためである。現実にHTSは創薬標的周辺やADME関連分子に対して行われるのみで、大半の生体分子に対する相互作用は測定されない。その結果、新薬は予期しない生体分子に結合して有害事象など様々な問題が後にヒトで生じ、市販後調査が必要になっていると考えることができる。
そういう観点で考えると、DRとは「見逃されている」薬理作用を介した未知の有効性を発掘しようという試みと言える。DRは様々な手法に基づき、ヒト病態iPS細胞を用いて既存薬のin vitroスクリーニングが行われたり、遺伝子発現において病態と薬物処置で反対方向に動くケースを元にして当該薬物の治療有効性を類推する、等が試されている(1)。確かにDRは低コストで安全性は担保できるが、有効性の確認を行う臨床試験が当然必要となり、既存薬の適応拡大ではメーカーも腰が重い。さらに臨床試験で有効性を立証できないという「死の谷」を時には渡れない現実がある。
本稿で紹介する私のアイデアは、もっと臨床予測性の高い低コストのDR創薬手法はないものだろうかという「問い」に端を発している。人間での薬物の有効性を予測するためには、やはり実臨床データを用いるべきだろう。薬学者に前向きな臨床試験はできない、でも後向き解析ならできる。そこへ2018年施行の次世代医療基盤法によって、ようやく我が国でも医療情報の研究活用に門戸が開かれた。臨床統計学や薬剤疫学で得た仮説を元にして、動物・細胞・分子レベルの基礎科学を展開するリバース・トランスレーショナルリサーチが今やっと可能になったのである。
そこで最初に目をつけたのは、有害事象レルフレポートであった。ここでは私がなぜ市販後調査の自発報告というバイアスの多いデータを使うことにしたのかを紹介するとともに、レギュラトリーサイエンスの将来への展望を考える意味でも電子化された医療情報が医療や創薬シーンを変える可能性について考えてみたい。
有害事象セルフレポートはヒト病態データベース
様々な医薬品は好ましくない副作用(有害事象)を引き起こす。有害事象を治験段階で網羅的に捕捉することは難しく、市販後調査によってモニターされ規制当局に報告される。米国FDAは国内外で発生した有害事象セルフレポートをデータベース化し、現在、1000万件もの有害事象発症例が世界中から誰もが見られる形で公開されている。これがFAERS(FDA Adverse Event Reporting System)である(2)。
有害な副作用のメカニズムの多くは、今なお不明のままである。一方、有害事象で見られる症状や病理は自然発症する疾患と一部で共通性を有することから、薬理学者はしばしば動物で病態モデルを作成するために有害事象を利用してきた。つまりFAERSは視点を変えると人間における貴重な病態誘発モデルと見なすことができる。しかもFAERS症例の約半数で多剤併用が行われているため、未知の薬物間相互作用が含まれる可能性が高い。
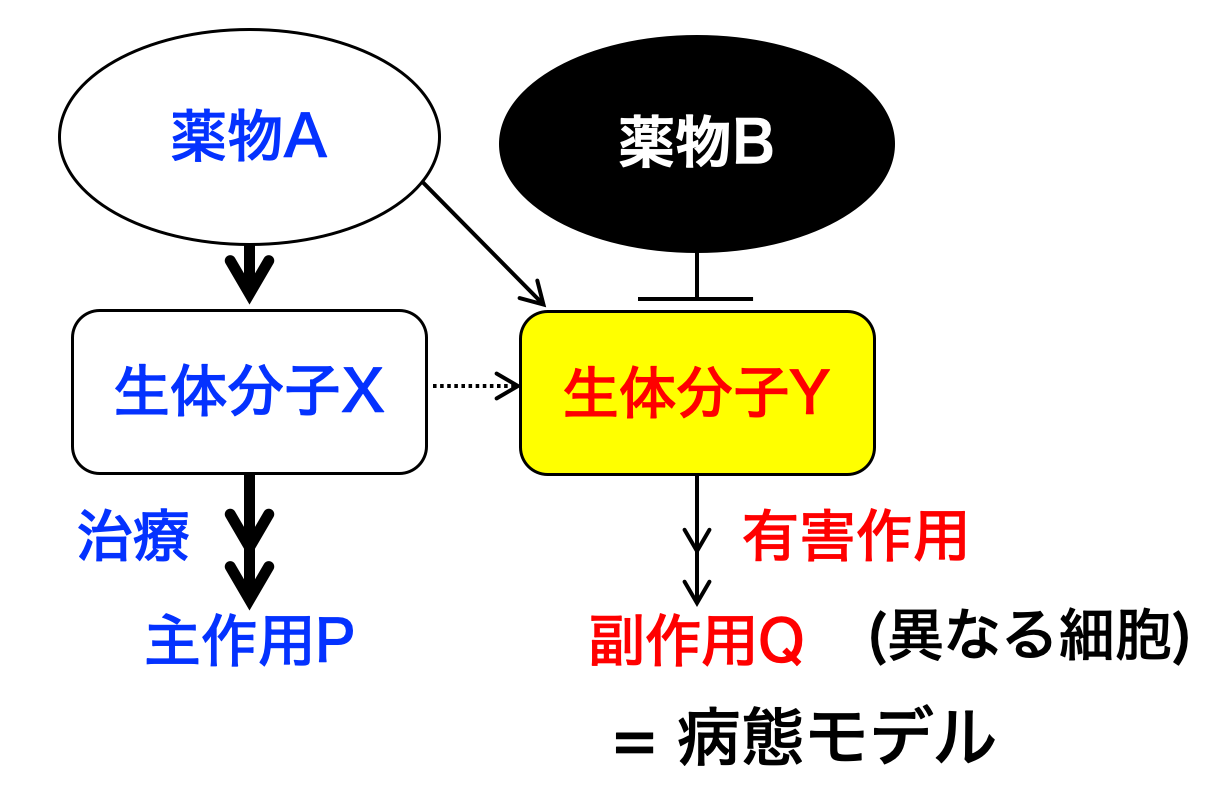
薬物Aが生体分子Xを介して治療に繋がる主作用Pを発揮するとき、有害事象Qは別の臓器や細胞にある異なる生体分子Yを介して発現すると考えることができる(右図)。例えば、中枢神経を興奮させるメタンフェタミン(覚醒剤)と抑制させるモルヒネ(オピオイド)はまったく異なる主作用点を有するにも拘わらず、結果的に報酬系ドパミンを放出させることで精神依存性という共通した有害事象を発揮する。
一方、ときに主作用Pと有害事象Qは共通した生体分子Xへの作用で起こり、その違いは細胞特異的な下流のシグナル経路に依存する。例えば、アスピリンによる抗炎症作用と胃潰瘍形成は共通のシクロオキシゲナーゼ阻害を介しているが、炎症細胞と胃では表現型が異なっている。
FAERSは人間で起こった有害事象例の蓄積であるから、これを薬剤誘発性疾患データベースであると見なすと、その発生リスクに影響する交絡因子を検討することは興味深い。当然、ADMEに影響する因子もヒットするだろうが、例えば多剤併用例を解析することによって、有害事象Qの発生率を低減させる偶然の併用薬Bを探索すれば、薬物Bは元々意図しない薬効によって薬物Aの有害事象Qを低減する治療・予防薬となるかもしれない。こうして有害事象セルフレポートの統計学的解析によってDRに至る仮説を導き出せる。
さらに発展させて有害事象メカニズムを追究するなら、薬物Aによる有害事象Qをそれ自体で研究する場合に比べたとき、併用薬BがQを抑制するという追加情報は未知の生体分子Yを特定する大きな手がかりとなる。こうして特定されたYは、有害事象の分子メカニズムであると同時に、有害事象に類似したヒト疾患の表現型に関与する分子であるかもしれない。もしそうなら、新たな創薬標的を創出することにつながり、新しい化合物を設計することによって知的財産を確保した創薬に繋げることができる。
臨床エビデンスに基づく仮説導出と実証実験プロセス
過去に報告した研究例(3)で用いた解析の手順を解説する(右図)。
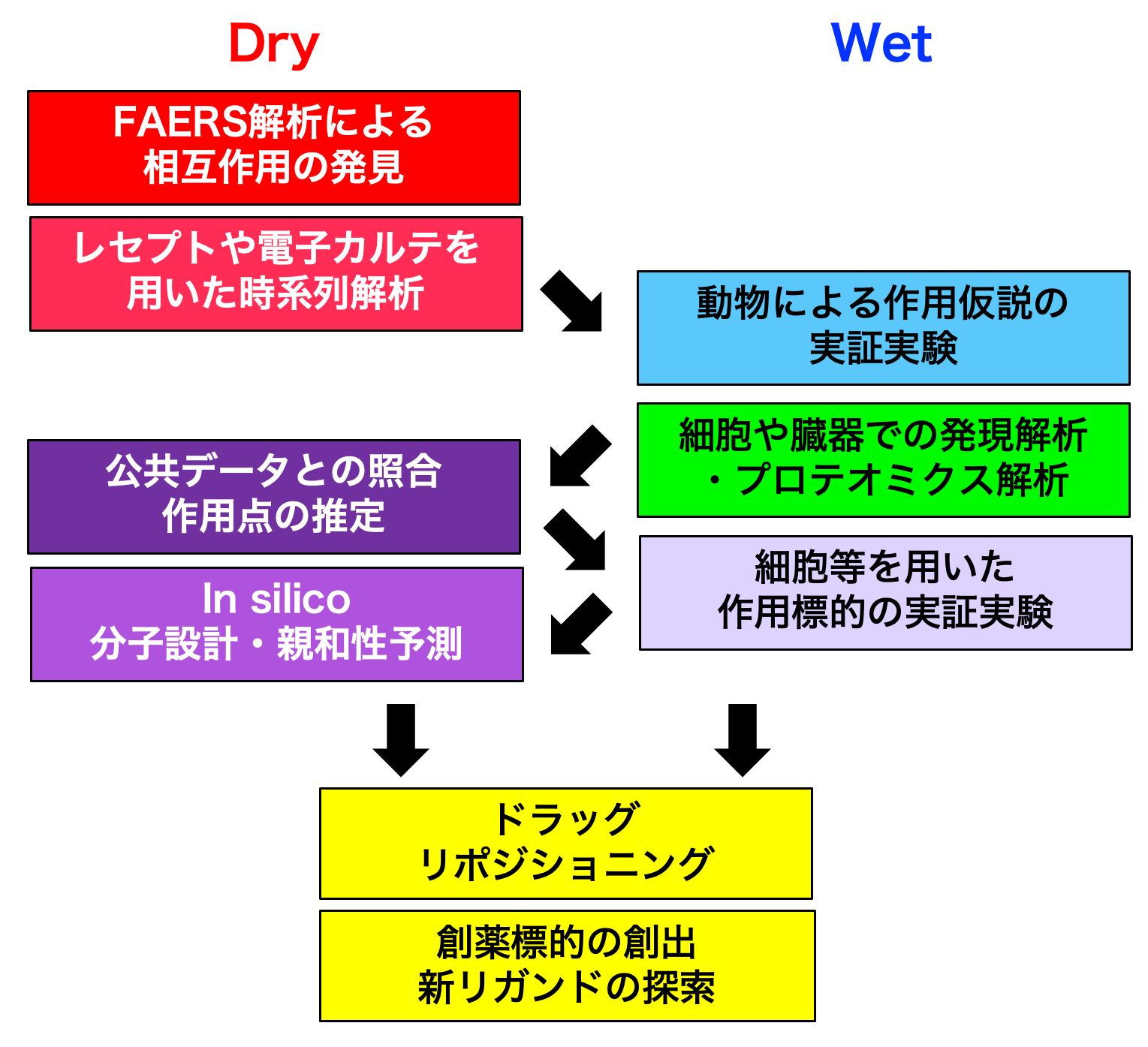
第1段階(Dry):データ解析による相互作用の仮説導出
我々はまず非定型統合失調症治療薬が起こす高血糖・糖尿病に着目してFAERS解析を行った。その結果、高血糖・糖尿病の報告率をクエチアピンが高いオッズ比で増大させていること、またその報告例数が数万件と十分に存在することを見いだした。次に層別解析によって、報告率の上昇が原疾患、性別、年齢等によって大きく左右されないことを確認した上で、クエチアピン使用例を母集団とした場合に用いられた併用薬のすべてについて、オッズ比を低減させる薬物を探索したところ、ビタミンDによって発生リスクが約3分の1に低下していることを見いだした。
現在では、このようにFAERSを用いて得られた仮説について、他の実臨床医療データ(Real World Data, RWD)を用いた仮説の検証を行うようにしている。JMDCレセプトデータベース(4)を用いると、患者の約4割が定期健康診断によって空腹時血糖やHbA1c値をモニターしているため、クエチアピン処方を受けた患者についてそれ以前のビタミンD使用の有無によって2群間に分けて血液検査値の時系列解析を行うことが可能である。
第2段階(Wet):動物や細胞を用いた実証実験
FAERS解析によって有望な薬物・薬物相互作用が検出された場合、実験動物や初代培養細胞などを用いてウェットでの実証実験を行い、仮説を確かめる。我々の例では、マウスへのビタミンD前投与がクエチアピン投与で誘発される高血糖と高インスリン血症を抑制することを見いだし、クエチアピンの有害事象が急性のインスリン抵抗性であると目測を立てた。このとき、標的臓器での遺伝子発現やプロテオミクス解析をしておくと公共データとの照合による作用メカニズムの推測に利用できる。
第3段階(Dry):公共ビッグデータを用いた作用メカニズムの仮説創出
動物や細胞に化合物を負荷した場合の遺伝子発現変動やリン酸化を網羅的に解析したデータが公共データベース、例えばGEO (5) など)に含まれている。先の実証実験で得られたデータとの照合などによって、作用メカニズムの仮説を得ることが可能である。我々の例では、クエチアピンによる遺伝子発現変動のトキシコゲノミクスデータからインスリン受容体下流に位置する生体分子に着目することになった。
第4段階(Wet):分子メカニズムの実証実験
有害事象メカニズムおよび併用薬の有益な作用について、作用メカニズム仮説に基づいた実証実験を設計、実施する。我々の例では、ビタミンDによるクエチアピン誘発高血糖の改善はインスリン受容体下流に位置するアダプタータンパク質PI3KR1の阻害とその発現上昇に起因すると結論づけた。
FAERSを用いるメリット・デメリット
FAERSに収録された報告数の多い薬物および有害事象を概観すると、医薬品としては古典的に頻用される有効成分に加えて近年使われるようになった抗体医薬や低分子薬が数多く並ぶ。一方、有害事象としては薬効欠如や適応外使用といった単純事象も含まれるが、全体に症状や徴候を表す概念が多い。これらは自発報告例に起因する特質の一部であり、次のような利点と欠点があることを十分に理解することが重要である。
利点①:例数が多く無料で使える匿名化RWDである
従来の疫学研究と比べて、FAERSが数百万例の症例を対象にできるRWDであることは最大の利点である。すでに匿名化がなされており、公開データをダウンロードすれば誰でも今日から研究対象とすることができる手軽さがある。Webで検索できるサイトAERSMineも制作・公開されている(6)。
利点②:世界中の多種多様な医薬品が用いられている
世界には日本で未承認あるいは知られていない有効成分が使われている医薬品が数多くあり、相互作用もそれに比例して多種多様である。FDAは米国内で医薬品を販売している企業に対して、当該薬物について世界中で発生した有害事象レポートの報告を求めているため、FAERSは日本を含め、実質的に世界中の医薬品についての有害事象例を広く収録している。
利点③:病名に拘わらず症候を捉えやすい
FAERSでの適応疾患や有害事象は世界的に統一されたMedDRA (7)によって記述されている。ICD-10 (8)などの病名体系とは異なりMedDRAは主観的な症状や徴候まで広く含まれていることから、永続的な病変に至らない一過性の有害事象を解析するのに適している。MedDRAには標準化されたクエリーSMQも備わっており、階層的な用語体系であるため、目的に応じた粒度の選択や用語の選択を行うことができる。この点は用語の標準化に多大なコストを必要とする他のRWDにはない利点である。
欠点①:重複症例が多数含まれている
自発報告は医薬品の製造者のみならず、医師や患者あるいは弁護士など多種多様な人々からなされるため、同一症例が重複して報告されるケースがある。またデータベースは年四半期ごとに更新されるため、同一症例の継続分には過去に報告された時点のデータが含まれている。これらは全体の10%以上にも及ぶため、データは事前にクリーニングする必要がある(9)。
欠点②:医薬品名が多種多様である
FAERSに報告された1000万症例では3700万の医薬品が使われており、その名称を単純に文字列として比較すると69万種類の名称が見いだせる。例えばモルヒネという化合物は塩を考慮しないと1種類であるが、様々な商品名や余分な記述の追加により2000種類ものバリエーションがFAERS中に見いだせる。この医薬品名を「名寄せ」する仕組みはFAERS自体にも一部備わっているが不完全であり、我々は独自開発しているライフサイエンス辞書(10)を用いて、69万種の表記を5000種類の有効成分名に98%以上の精度と網羅率で名寄せしているが、これは13,000種のルールとスペルミスを修正する機械学習を用いたテキスト処理で実現している。
欠点③:様々なバイアスや偽陽性シグナルを考慮する必要がある
市販後すぐの医薬品は注目を浴びることから報告数が多くなり、時間とともに報告数は減少する。また、安全性情報が規制当局から公表されると当該薬あるいは類似薬について報告数が増加する。このような人的なバイアス(11)があるため、FAERSで有害事象発生率は正確に評価できない。また、ある薬物について特定の有害事象報告が多いと、他の有害事象シグナルは弱くなる。反対に、ある有害事象について特定の薬物で報告が多い場合、他の薬物のシグナルは弱くなる。これはcompetition biasと呼ばれる。
さらにFAERS解析で生じやすいのは偽陽性シグナルである。これは治療目的で使用した薬物が薬効不十分で有害事象に適応症が記載される場合(例:癌性疼痛に対してモルヒネを用いた症例における「疼痛」関連の有害事象)や、一般的によく併用される薬物に見かけ上の有害事象軽減効果が見られてしまう場合などが挙げられる。
今後に向けて
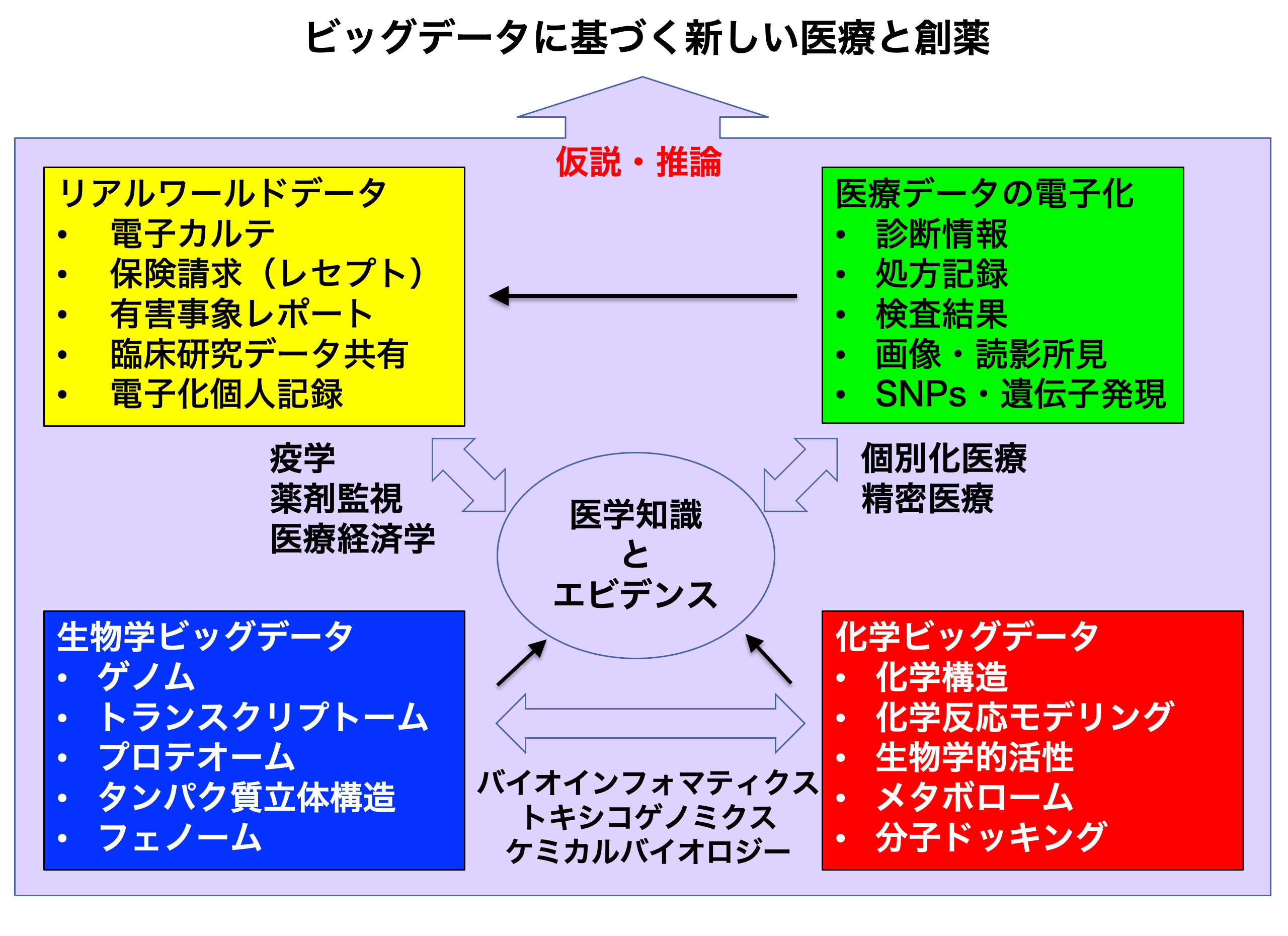
これまでの生物学や化学は、すでに膨大なデータの蓄積を重ね、それらの多くは共有されて研究に利用されるとともに医学知識と臨床エビデンスの構築にも貢献してきた(右図)。一方、臨床において個別化医療や精密医療の推進に医療情報を活用する期待は高まってはいるものの、電子カルテなどの実診療行為を中心としたRWDの共有と利活用は今ようやく始まったところである。
欧米で先行した医療情報化は、RWDを中心として医療ニーズの可視化や臨床エビデンスの改良に用いられるなど、創薬にも影響を及ぼしつつある。本稿で紹介した手法も単にFAERSの活用に留まらず、RWDの活用に発展することで、多角的に創薬シーンへの展開が可能になる。動物や細胞あるいは分子レベルでの作用から臨床へのトランスレーションを基本としてきた創薬科学も、今後は臨床エビデンスに基づく「data-driven」アプローチで臨床予測性の高い作業仮説を生成することによって、新しい展開が期待できるだろう。
しかしRWDを扱い始めるとすぐに気づくのは、電子化された医療情報の大半が統制化されていない用語での記述に基づいており、その標準化やデータの構造化に多大な労力を有することである。その意味でも有害事象セルフレポートがMedDRAという体系化されたシソーラス(同義語辞書)で記述されていることは貴重である。
現在、電子カルテや看護レポートに記述されているテキストを自然言語処理によって匿名化と同時に構造化し、今よりもさらに深化したRWDの利活用を行おうとする動きが各方面で始まっている。こういったテキストの機械可読化処理には自動翻訳と同じくニューラルネットワークを模した深層学習が極めて有用であり、専門家がアノテートした教師データを元にして人間が下すような判断を人間と同等以上の精度で自動的に行うことが可能になる。
例えば1つの応用例としては市販後調査が挙げられる。現在のように人手をかけて情報を収集し、その情報からドキュメントを制作して報告するスタイルを脱却して、電子カルテの機械的解読によって医薬品の有害事象発生例を自動的に検出する、いわゆる人工知能が実現化して市販後調査に省力化が図られる時代も、そう遠い話ではないだろう(12, 13)。そうした未来に備えて、人類の英智である医学用語を体系化したシソーラス、そして例えば医薬品とその適応症や有害事象との関係性を機械可読な形式でグラフ化したオントロジーの構築と整備が期待される。
文献リスト
- Pushpakom, S.; Iorio, F. Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNameee, C.; Norris, A.; Sanseau, P.; Cavalla, D.; Pirmohamed, M., Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov., 2019, 18, 41-58.
- FDA Adverse Event Reporting System
- Nagashima, T.; Shirakawa, H.; Nakagawa, T.; Kaneko, S., Sci. Rep., 2016, 6, 26375.
- JMDC Claims Database
- Gene Expression Omnibus
- Sarangdhar, M.; Tabar, S.; Schmidt, C.; Kushwaha, A.; Shah, K.; Dahlquist, J.E.; Jegga, A.G.; Aronow, B.J., Data mining differential clinical outcomes associated with drug regimens using adverse event reporting data. Nat. Biotech. 2016, 34, 697-700. AERSMine
- MedDRA
- ICD-10
- Banda, J.M.; Evans, L.; Vanguri, R.S.; Tatonetti, N.P.; Ryan, P.B.; Shah, N.H., A curated and standardized adverse drug event resource to accelerate drug safety research. Sci. Data, 2016, 3, 160026.
- ライフサイエンス辞書
- Weiss-Smith, S.; Deshpande, G.; Chung, S.; Gogolak, V., The FDA drug safety surveillance program: adverse event reporting trends. Arch. Intern. Med., 2011, 171, 591-593.
- Li, F.; Liu, W.; Yu, H. Extraction of information related to adverse drug events from electric health record notes: design of an end-to-end model based on deep learning. JMIR Med. Inform., 2018, 6, e12159.
- Basile, A.O.; Yahi, A.; Tatonetti, N.P. Artificial intelligence for drug toxicity and safety. Trends Pharm. Sci., 2019, 40, 624-635.

