|
Research
Research Projects
We have advocated elucidating the cellular functions through the measurement of biomolecules based on analytical chemistry. In particular, we have focused on proteome science consisting of mass spectrometry, nano-separation science, computational science and cell biology to develop the methodologies for the functional analysis of cells. More specifically, we are conducting research on the following five topics;
- Development of novel analytical technologies for proteomics
- Human proteome analysis based on single-shot LC-MS systems
- Elucidation of intracellular phosphorylation network analysis
- Quantitative clinical proteome analysis of tissue samples
- Studies on the molecular targeting drug discovery based on phosphoproteomics
Unlike genomic and transcriptomic researches, proteomics is still immature in terms of the measurement technologies and the complete analysis of proteome has not been established yet. The final goals of proteomics are to uncover the cellular protein events such as (1) protein expression/degradation, (2) protein localization, (3) protein interaction, (4) protein post-translational modifications (PTM) and (5) protein processing/splicing in proteome-wide.
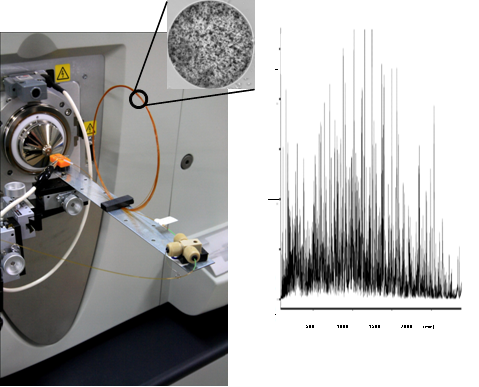 Figure 1 Complete proteome analysis by nanLC-MS.
Figure 1 Complete proteome analysis by nanLC-MS.
(Left) nanoLC-MS system with 3.5 meter monolithic silica column.
(Right) Total ion current chromatogram of E. coli proteome. Analysis on a microarray scale was achieved.
We are aiming to develop novel approaches to tackle the technical barriers and to explore proteomic researches for clarifying the biological problems.
In order to analyze the entire proteome expressed in cells, we are focusing on developing efficient separation systems based on nanoLC-MS using meter-long monolithic silica capillary columns with the world's highest performance beyond theoretical plate number 1,000,000. So far, our systems allowed to expand the measurable dynamic range of highly complex proteomics samples, achieving the analysis of Escherichia coli expressed proteome on a microarray scale (see Figure 1). This system is currently applied to more complex proteome such as human. We are also developing new technologies for quantitative proteomics as well as high sensitivity proteomics.
In cellular signal transduction network, reversible phosphorylation is one of the key events to transduce the signal into nucleus to control the gene expression. Approximately 30% of human proteins were estimated to be phosphorylated. We have developed a highly selective enrichment method for phosphopeptides and applied to proteome-wide acquisition of cellular phosphorylation status. Consequently, we found that at least 70% of human proteins are phosphorylated, which are 2-fold more than that registered in the public protein database such as UniProt. The next step is to intertwine the kinases with their substrates for revealing the whole picture of signaling network by using experimental and computational approaches.
Our phosphoproteomics system has been also employed to carry out in vivo phosphoproteome profiling of the molecular-targeting drugs, which would facilitate drug discovery for cancer therapy. Furthermore, we are exploring the functional analysis of newly discovered phosphorylation molecules. In addition to phosphorylation, we recently started other PTMome analysis to evaluate the cellular and molecular functions.
Recent publications
- Iwasaki et al., One-dimensional capillary liquid chromatographic separation coupled with tandem mass spectrometry unveils the Escherichia coli proteome on a microarray scale. Anal. Chem., 82, 2616-20, 2010.
- Imamura et al., Towards the systematic discovery of signal transduction networks using phosphorylation dynamics data. BMC Bioinformatics 11(1), 232, 2010.
- Sugiyama et al., Phosphopeptide enrichment by aliphatic hydroxy acid-modified metal oxide chromatography for nano-LC-MS/MS in proteomics applications. Mol. Cell. Proteomics 6, 1103-9, 2007.
- Ishihama et al., Quantitative mouse brain proteomics using culture-derived isotope tags as internal standards. Nat. Biotechnol. 23, 617-21, 2005.

|