
Research
Synthetic organic chemistry is a fundamental science and technology to create materials, drugs, and any other organic compounds which bring a better life. A key for the progress in this research field is the development of new synthetic methods for making use of highly reactive chemical species for the synthesis of useful molecules. To address this mission, our research group focuses on the rational design of unprecedented catalysts and reagents that lead to the establishment of groundbreaking new methodologies.
Current Research
Development of New Methods for Generation of Organoradicals
Radical species show attractive features, distinguished from ionic species (i.e., anions and cations) in terms of the reactivity and selectivity, and hence they can provide a powerful tool in organic chemistry. On the other hand, radical chemistry has always lagged behind ionic chemistry, and has not yet been developed to a synthetically useful level due to the lack of practical approaches for the generation of radical species. To fill the blank, we developed the following new methodologies for the generation of various radical species under mild conditions.
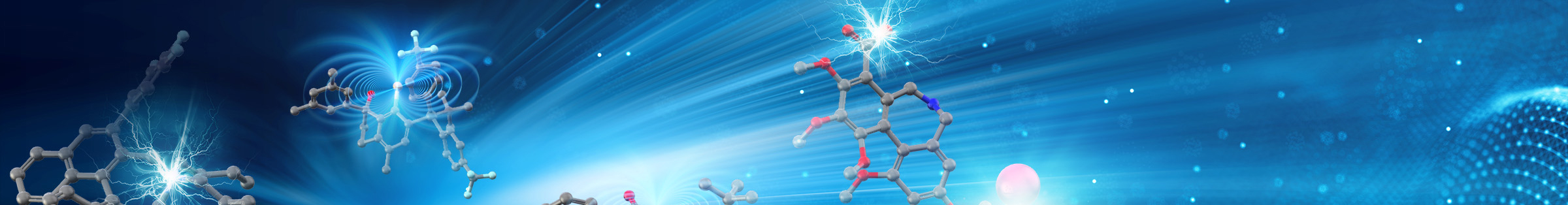
Alkylsilyl Peroxides as Efficient Alkyl Radical Precursors
Organic peroxides such as DTBP (di-tert-butyl peroxide) have been widely utilized as alkyl radical precursors. However, this method can provide only methyl radical or other structurally simple alkyl radicals under harsh reaction conditions. Recently, our group discovered that alkylsilyl peroxides (ASP), which have silyl group directly connected with peroxide structure (-O-O-), can be readily prepared and easily handled as bench-stable alkyl radical precursors. With these reagents in hand, a range of alkyl radicals bearing functional groups can be generated in the presence of appropriate transition-metal catalysts under mild reaction conditions. Notably, this advantage allows for several attractive applications such as the construction of complex carbon skeletons and highly regio- and stereoselective carbon–carbon bond formation.
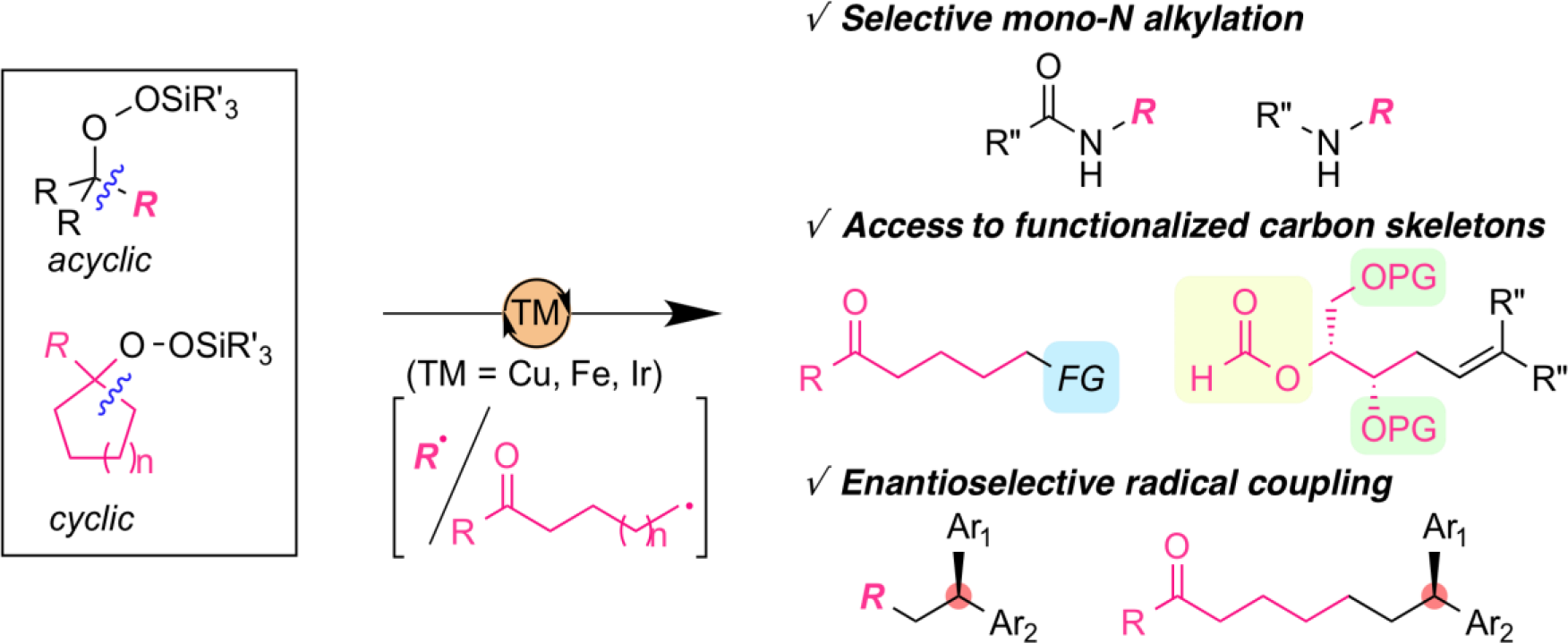
Design of Novel N-oxyl Radical Catalysts for C–H Functionalizations
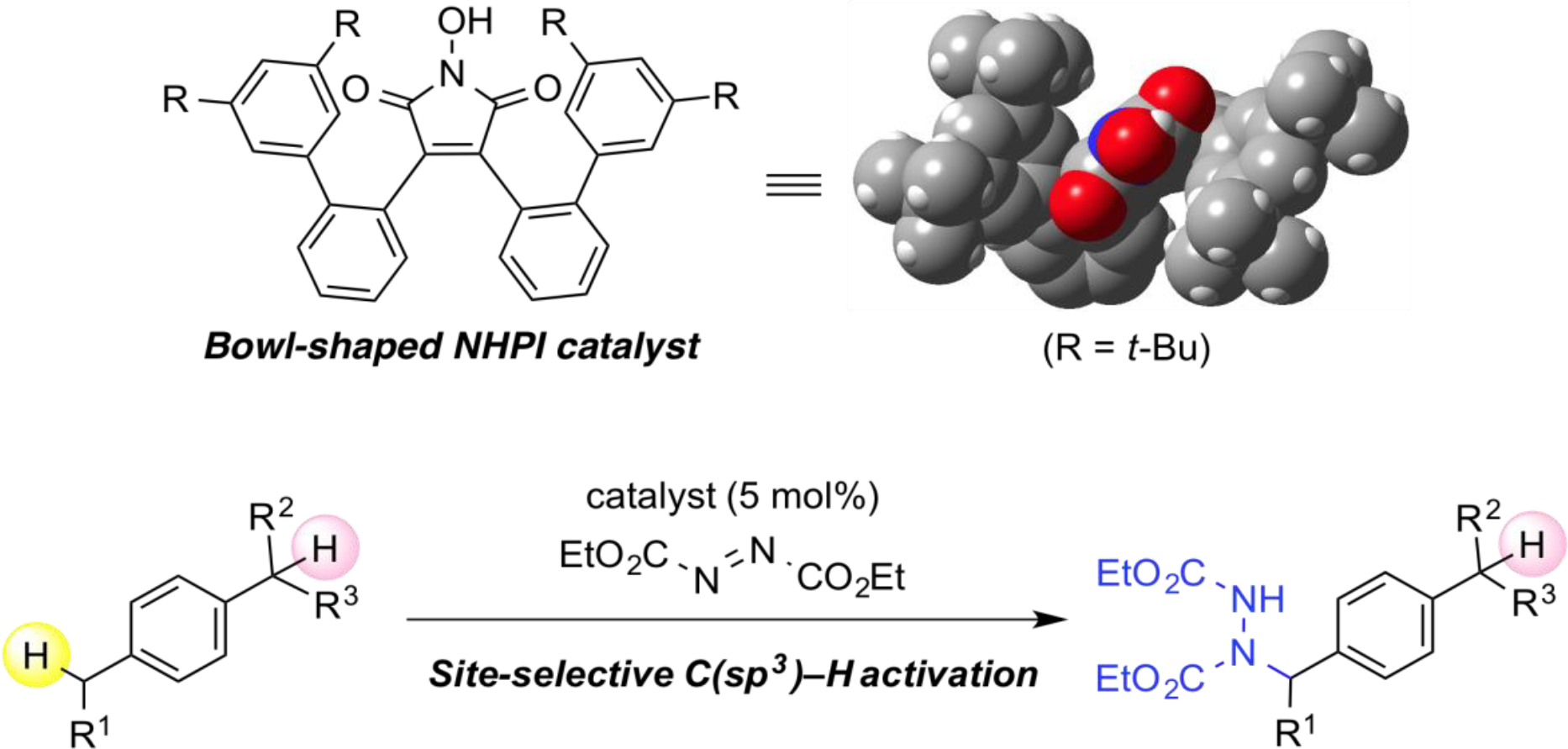
The development of selective and direct functionalization of C–H bonds is one of the most active research topics in organic chemistry as these strategies can provide high value-added chemicals from readily available feedstocks. The hydrogen atom abstraction of C–H bonds by N-oxyl radicals is one of the efficient methods for the generation of alkyl radicals from simple substrates. One drawback of this method is the lack of structural diversity for N-oxyl radicals, which has been limited their applicability for C–H functionalization reactions. In order to exploit their potential as practical organoradical catalysts, we developed novel N-oxyl radicals based on original molecular design and applied them into distinctive C–H functionalization reactions.
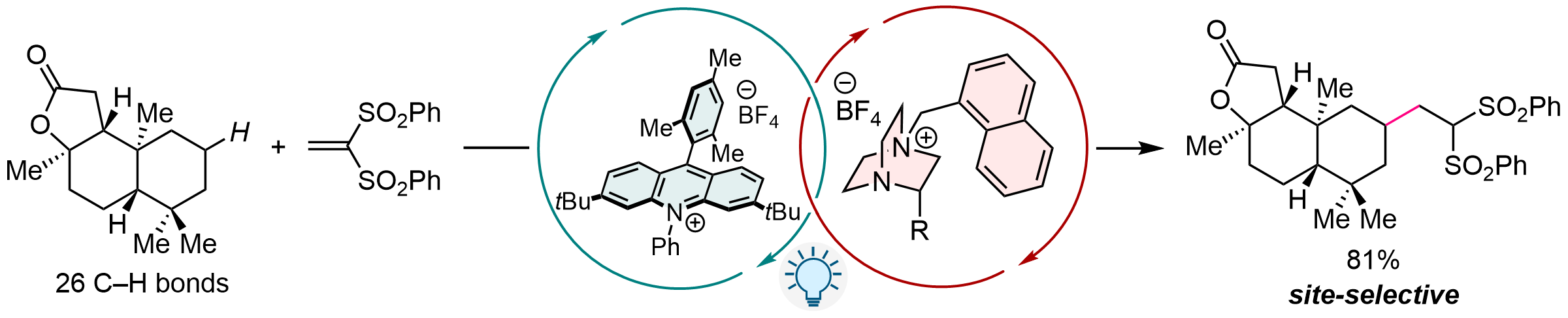
Cationic DABCO-Type Catalysts for Photoinduced Hydrogen-Atom Transfer
Photoinduced hydrogen-atom transfer (HAT) catalysis has attracted attention as a promising method for the C-H functionalization under mild conditions with high atom efficiency. Although various HAT catalysts have been developed, few of them are applicable to aliphatic compounds with strong C-H bonds and in most cases show insufficient site selectivity. We have developed new HAT catalysts that are readily accessible from inexpensive 1,4-diazabicyclo[2.2.2]octane (DABCO), and have achieved site-selective C-H alkylation of unactivated substrates using them with an acridinium-type photoredox catalyst under visible light irradiation.
Research on Organocatalytic Chemistry
Research Direction
Based on the information and research findings on the design of fine Lewis acids and organometallic catalysts, the rational design of high-performance organocatalysts will be executed in the research project, which is divided into four main research items consisting of "organobase catalysts", "organoacid catalysts", "organoacid/base catalysts" and "organoradical catalysts". Throughout both basic and applied researches in this project, we would like to aim at the design and synthesis of a series of truly high-performance organocatalysts for practical organic synthesis including asymmetric transformations.
Organobase Catalysts
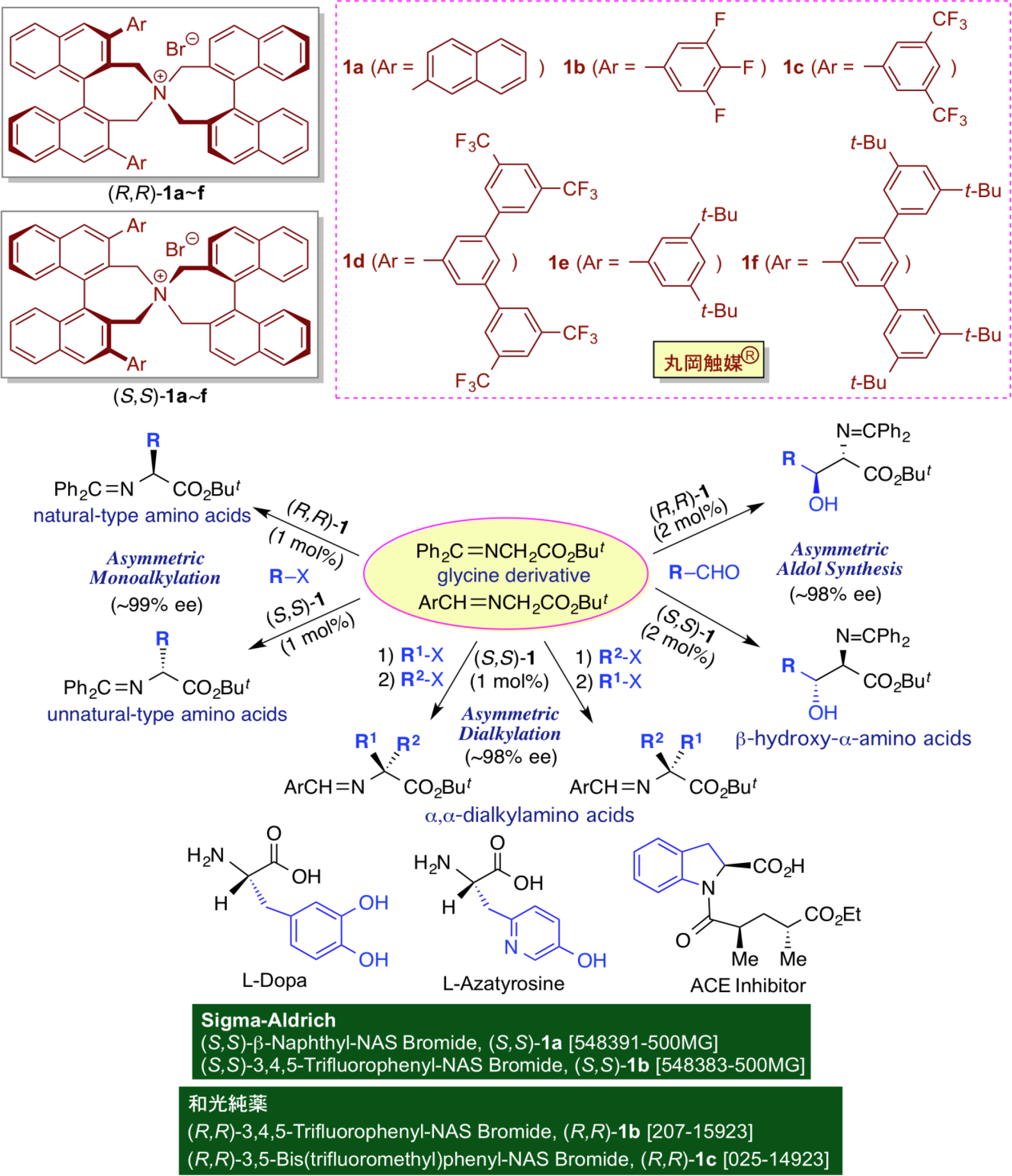
As an organobase catalyst, we focus on the design of chiral phase-transfer catalysts, and conducted the following basic and applied researches. The tetraalkylammonium salts (R4N+X-) as phase-transfer catalysts are usually water-soluble due to its ionic structure. However, when the alkyl group becomes a longer chain, it becomes more lipophilic and soluble in organic solvents. By utilizing this unique property as a phase-transfer catalyst, various phase-transfer reactions can be carried out at room temperature and in the presence of water, and at the same time, a large increase in the reaction rate can be expected. Furthermore, there are various advantages on organic synthesis such as the simple experimental operation and the ease of large-scale reactions. From this viewpoint, the phase-transfer reaction can be carried out in an aqueous solution at room temperature, under atmospheric pressure with open system, thereby indicating that the industrialization of the phase-transfer reaction system is extremely easy. Unfortunately, however, it was not so easy to design and prepare an efficient chiral phase-transfer catalyst for asymmetric synthesis. The only successful examples so far have been the chiral phase-transfer catalysts derived from cinchona alkaloids, but these are always limited for efficient organic synthesis by the catalyst design and the use of halogenated solvents. In order to find conceptually new, chiral phase-transfer catalysts for various practical asymmetric transformations, we introduced commercially available, optically active (R)- and (S)-binaphthol, and newly prepared chiral spiro-type phase-transfer catalysts, (R,R)-1 and (S,S)-1, which are known as "Maruoka Catalyst®", and commerciallized as reagents from Sigma-Aldrich Co. LLC and Wako Pure Chemical Industries. Since then, we have been developing various asymmetric phase-transfer reactions under the influence of our chiral phase-transfer catalysts. Among these, the asymmetric alkylation of glycine derivatives using Maruoka Catalyst® enables the highly efficient, practical synthesis of various optically active, natural- and unnatural-type α-amino acids and their related products including optically active amino aldehydes, amino ketones, amino alcohols, etc. For example, therapeutic agents for Parkinson's disease, L-DOPA, antibiotics, L-azatyrosine, ACE inhibitors, etc. can be easily synthesized as physiologically active amino acids at a practical level.
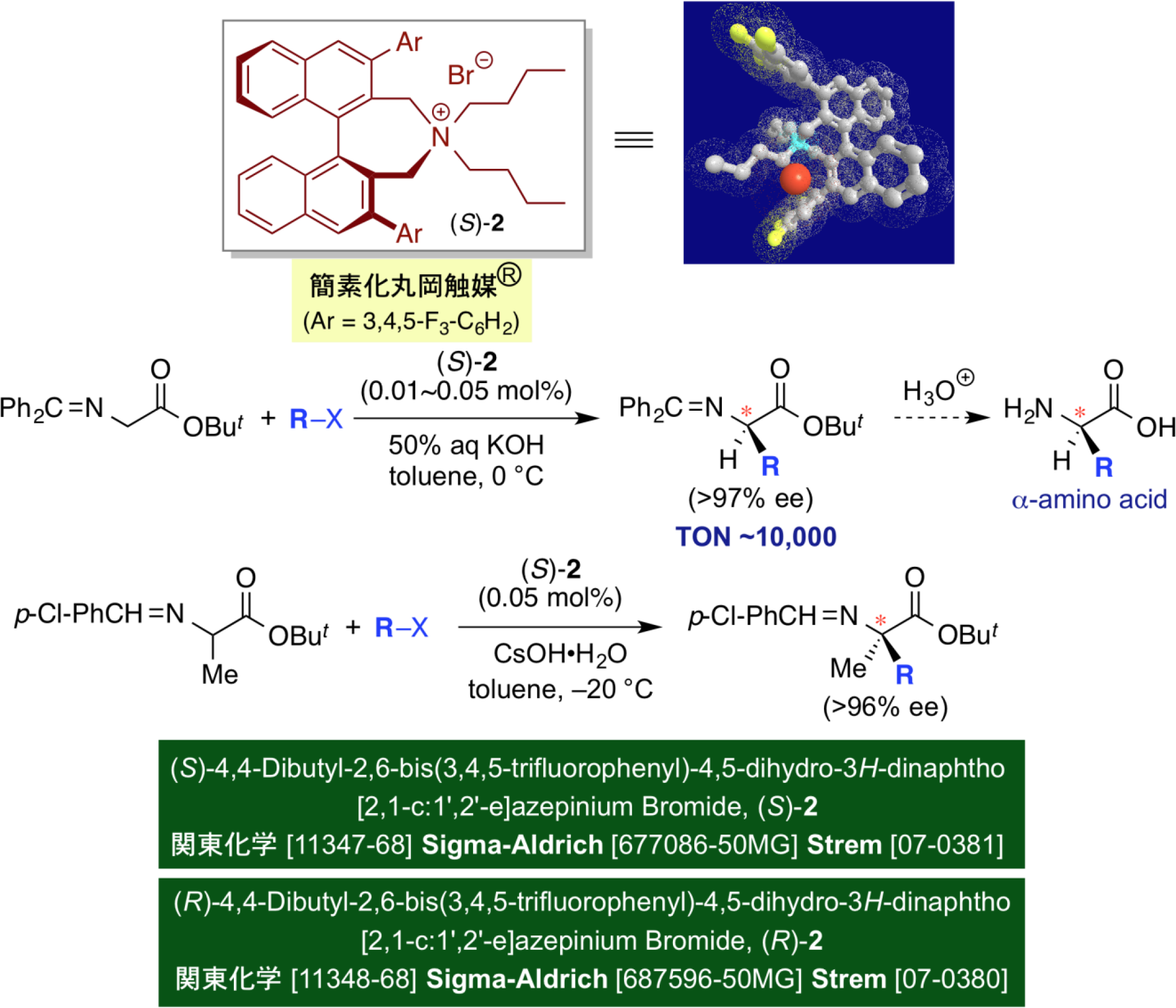
With the aim of further industrializing artificial amino acid synthesis utilizing our chiral phase-transfer catalysts, we focus on the design of chiral high-performance phase-transfer catalysts with the simplification of the catalyst structures. Ideally, the use of one binaphthyl moiety is highly desirable instead of using two spiro-type binaphthyl groups. Accordingly, we extensively studied on the design of new, yet simplified phase-transfer catalysts with one binaphthyl core, and by introducing a combinatorial design approach, we finally found that the catalyst of type (S)-2 is among the best. Indeed, the asymmetric alkylation of glycine derivatives can be effected very smoothly with extremely low catalyst loading, i.e., in the presence of only 0.01 mol% of (S)-2, with excellent enantioselectivity. Moreover, by applying this method to α-alkylamino acid derivatives, a catalytic asymmetric synthesis of optically active α,α-dialkylamino acids has been developed as well. With respect to this chiral catalyst, we acquired the registered trademark "Simplified Maruoka Catalyst®" and now it is commercially available as a reagent from Sigma-Aldrich Co. LLC, Strem Chemicals, Inc. and Kanto Chemical Co., Inc. By utilizing this super-active catalyst, we succeeded the industrialization on the eficient synthesis of various artificial amino acids as valuable pharmaceutical intermediates by Nagase & Co. Ltd. and Kishida Chemical Co., Ltd.

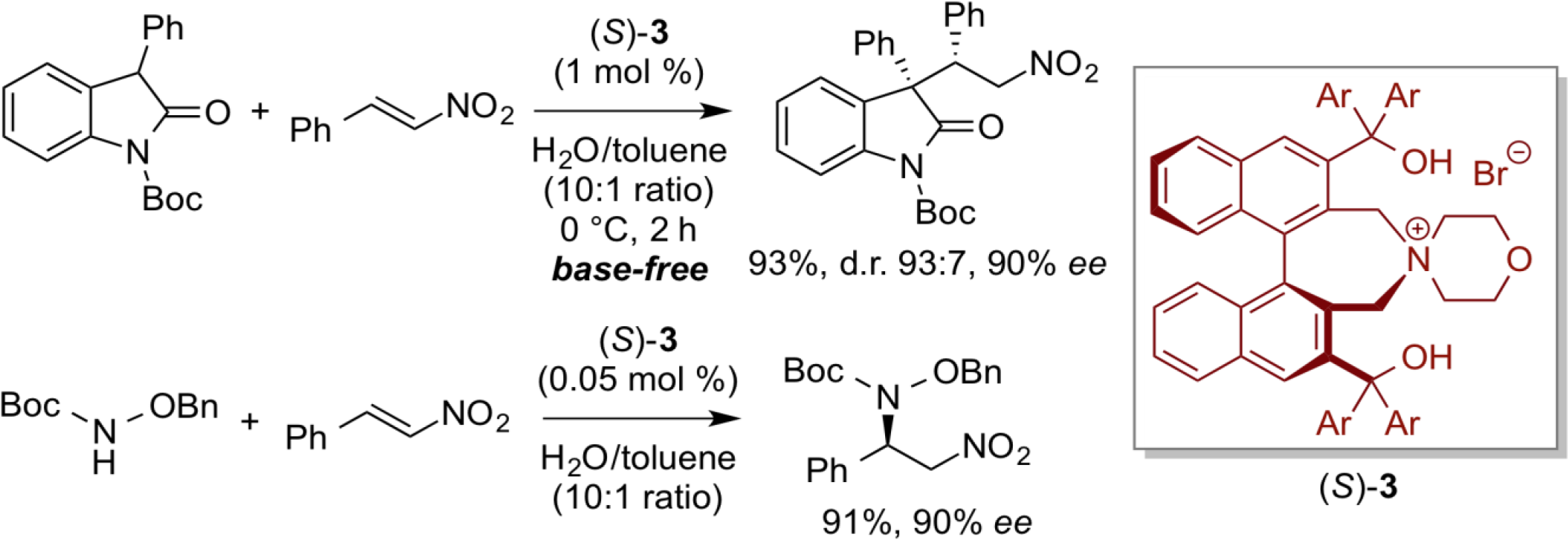
In the reaction system using a quaternary ammonium salt as phase-transfer catalyst as described above, it has been considered that addition of a base such as potassium hydroxide is essential for promoting the phase-transfer reactions. However, it is possible to find a chiral phase-transfer catalytic reaction system that proceeded under neutral conditions without using any external base. In this reaction system, the phase-transfer reaction can be carried out in the presence of chiral bifunctional phase-transfer catalyst of type (S)-3 by adding a very small amount of an organic solvent for dissolving organic substrates and the catalyst in an aqueous solvent. Hence, this neutral reaction condition consists of truly an environmentally-benign, practical reaction system.
Organoacid Catalysts
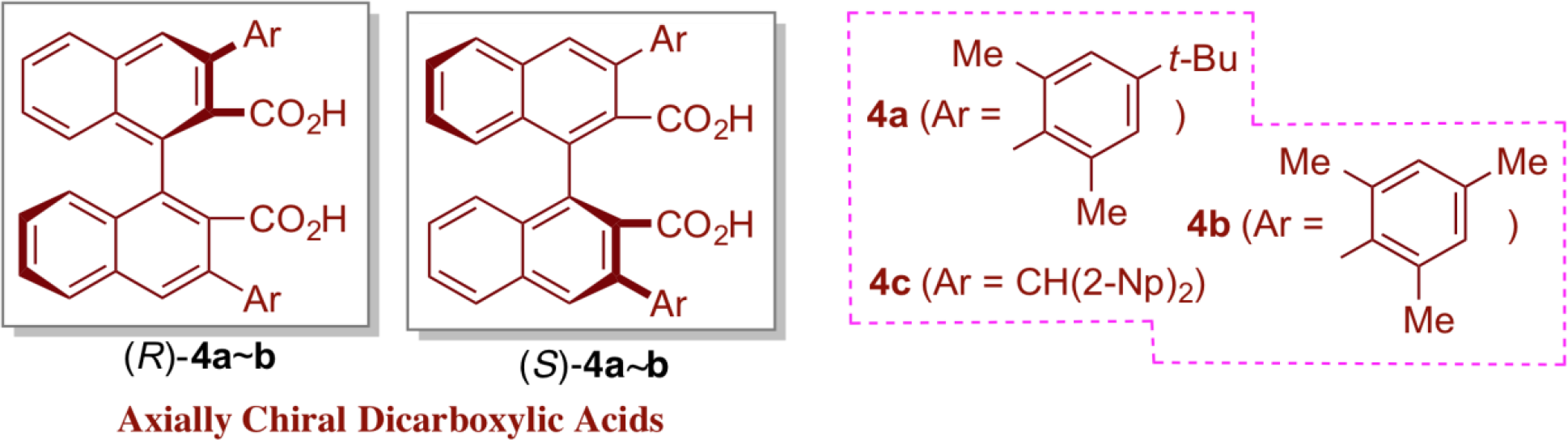
In catalytic asymmetric synthesis using a chiral organoacid catalyst, the acidity of the hydrogen bond donor is the key to the realization of the reaction. The necessity to use them properly arises from the choice of optically active alcohols, thioureas, and phosphoric acids. Interestingly, however, there have been only few reports using carboxylic acids, which are the most common weak acids, as organoacid catalysts. This is in sharp contrast to the fact that proline, which is the most important and highly practical organic acid/base catalyst, utilizes a carboxylic acid as its acid catalyst site. The difficulty of using carboxylic acids in the research of organoacid catalysts is attributed to the following two points: (1) difficulty in constructing an effective asymmetric environment around carboxylic acid moiety, and (2) low reactivity of carboxylic acid as an acid catalyst. In order to overcome these difficulties, we introduced the axially chiral dicarboxylic acid catalyst of type 4, which arranges two carboxylic acids at a distance that allows intramolecular interaction via hydrogen bonding, thereby exhibiting a unique character that is clearly different from other organoacid catalysts.
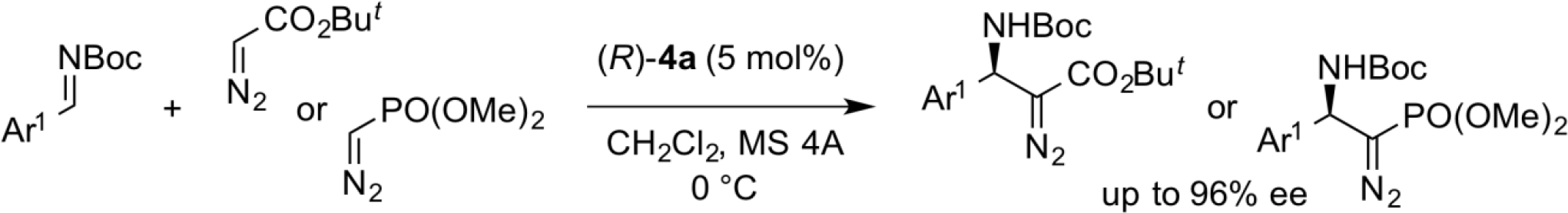
Asymmetric Mannich-Type Additions of Diazo Compounds
We have found that diazoacetic acid ester and diazophosphoric acid ester reacted with N-Boc imines under the influence of axially chiral dicarboxylic acid catalyst (R)-4a to furnish the corresponding Mannich-type products with high enantioselectivity. The optically active -amino--diazo esters, thus obtained, are easily converted into the corresponding β-amino acids and aminophosphates, which are useful as pharmacologically active intermediates.
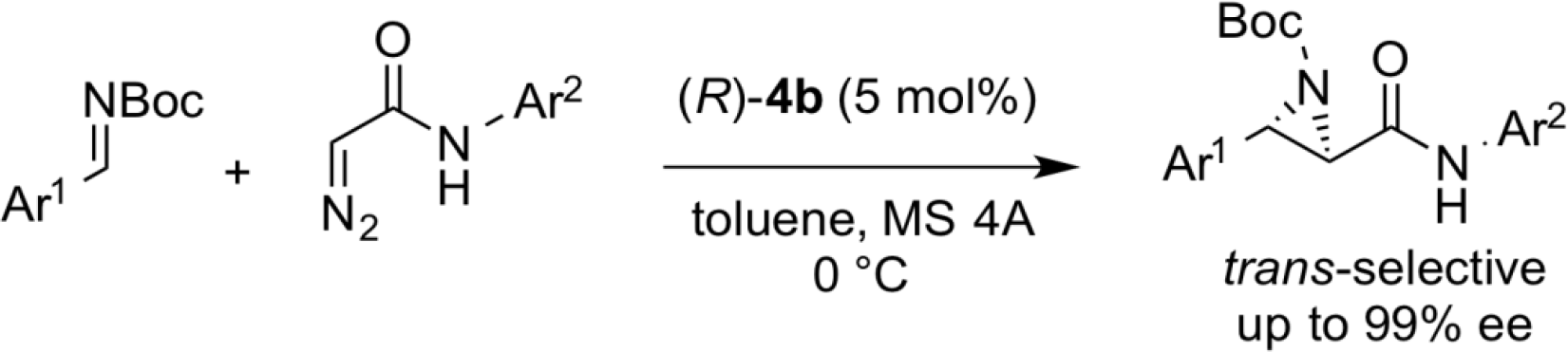
trans-Selective Asymmetric Aziridination
In the Mannich-type reaction as described above, switching diazoacetates to diazoacetamide as substrate, it was clarified that the aziridination reaction proceeded selectively. While conventional chiral Lewis acid catalysts exhibited cis-selectivity, the present reaction afforded the trans isomer preferentially.
Asymmetric Imino-Azaenamine Reactions
We have also developed a catalytic asymmetric addition of azaenamine, derived from the corresponding N,N-dialkylhydrazine and aldehyde, with N-Boc imine as shown below. This asymmetric reaction is characterized by excellent stereoselectivity and substrate generality that are not achievable by existing catalyst systems.
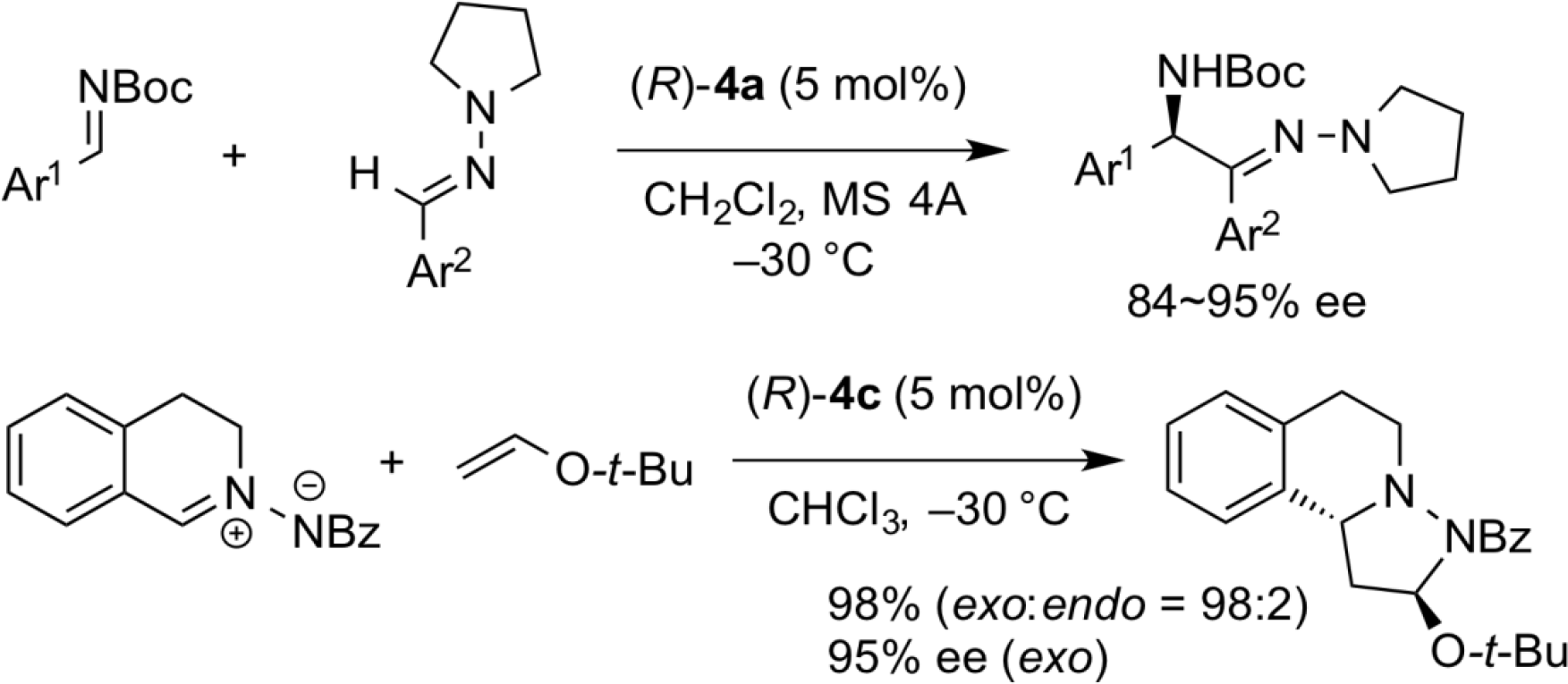
Chemistry of Boron-Containing Organoacid Catalysts
The chiral acid catalyst (S,S)-7, which was prepared from a chiral diol 5 and 2-boronobenzoic acid 6, exhibited unique reactivity, and it was clarified that the trans-selective asymmetric aziridination reaction of N-Boc imine with diazoacetamide is realized with high diastereo- and enantioselectivity.
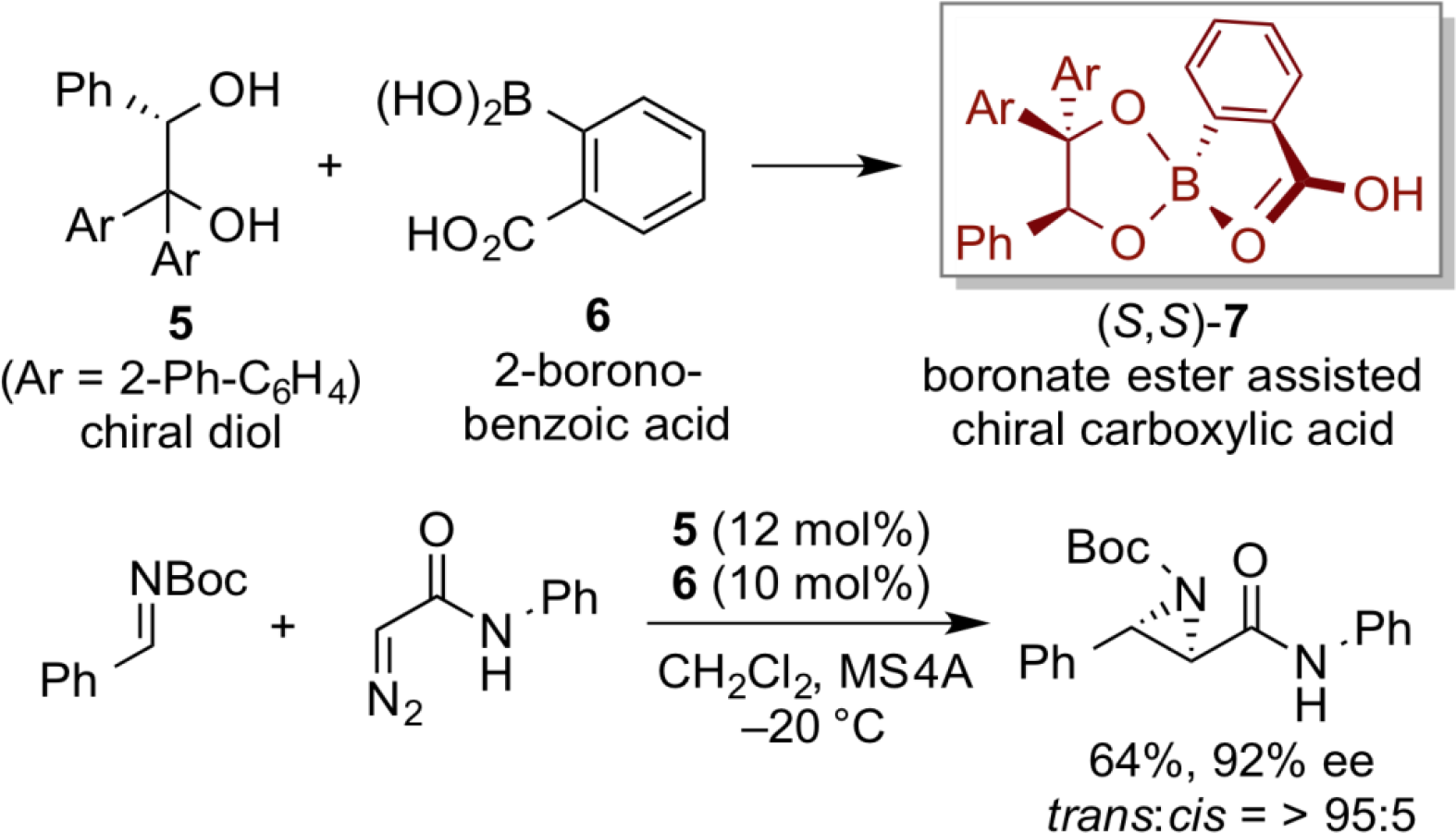
Another chiral organoacid catalyst (S)-8 was designed based on the use of chiral boronic acid possessing optically pure binaphthol, and highly enantioselective aza-conjugated addition of hydroxamic acid with quinone imine ketal has been developed by using (S)-8 catalyst.
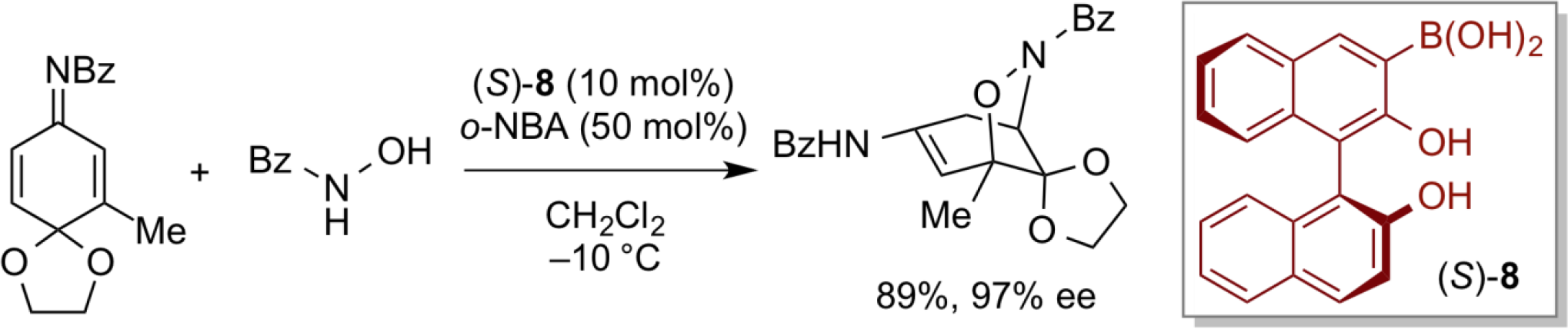
Furthermore, in recent years, as a totally new research development, we have succeeded in designing a new electrophilic selenium species, which is regarded as a highly efficient chiral organic π-acid catalyst 9. This electrophilic selenium-type chiral π-acid catalyst 9 can be successfully applied to asymmetric lactonization of β,γ-unsaturated carboxylic acids with excellent enantioselectivity.
Organoacid/Base Catalysts
The organocatalysts, that have been extensively studied in recent years, are often derived from natural sources such as proline and other amino acids and cinchona alkaloids, and have several advantages such as ready availability of starting materials and easy preparation of organocatalysts. So far, its efficiency has been shown in various asymmetric reactions. On the other hand, however, since the structure of the catalysts strongly depends on the structure of the natural products as starting materials, there is a limitation on the rational design of organocatalysts for the intended reactions. Accordingly, we have introduced a chiral binaphthyl moiety, which is an artificial asymmetric element with ready availability and easy modification, as a core structure of the catalyst. The precise design of chiral organocatalysts of types 10, 11, and 12 by our strategy would exhibit unique reactivity and selectivity, not observable by the conventional organocatalysts.
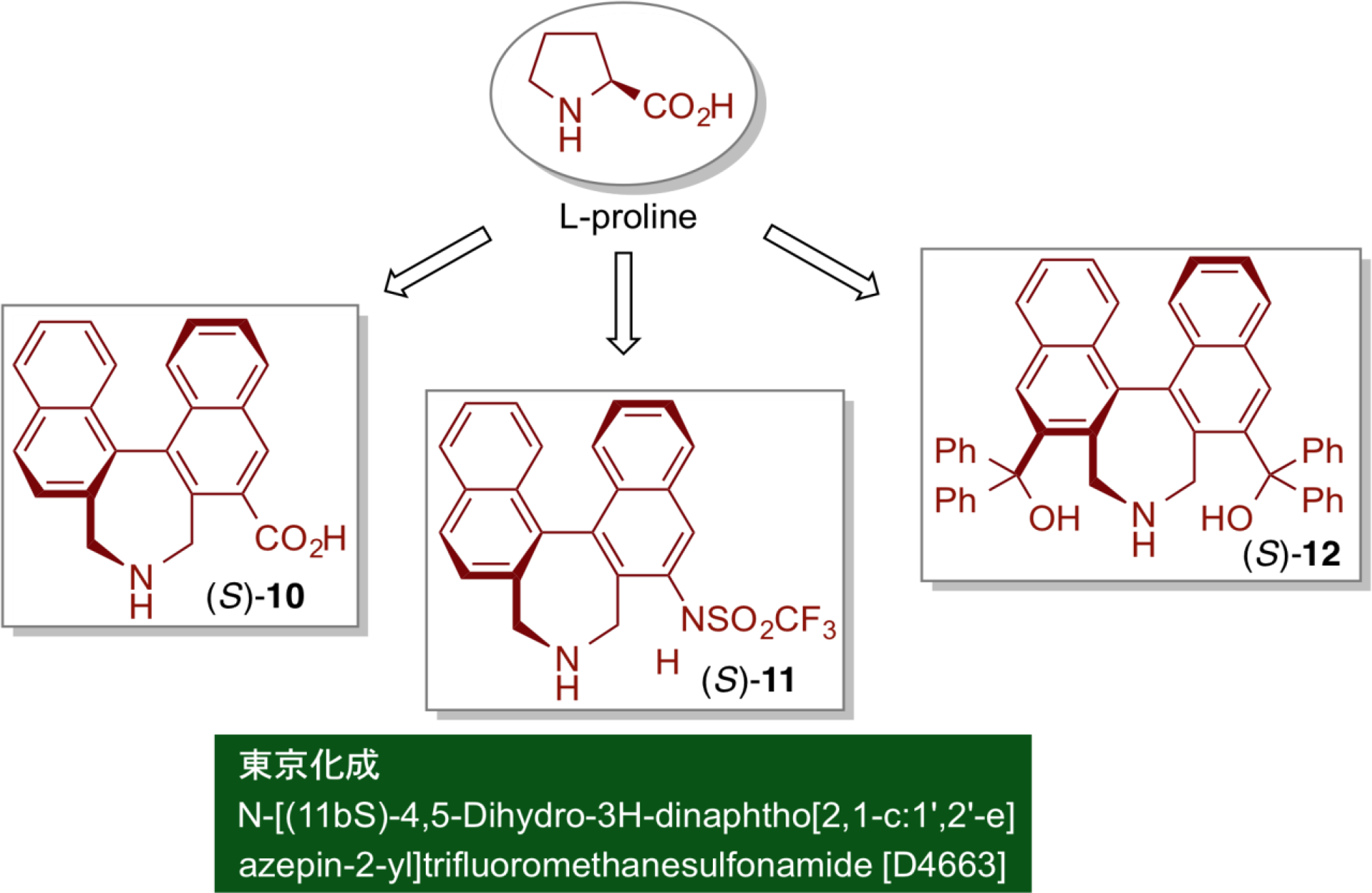
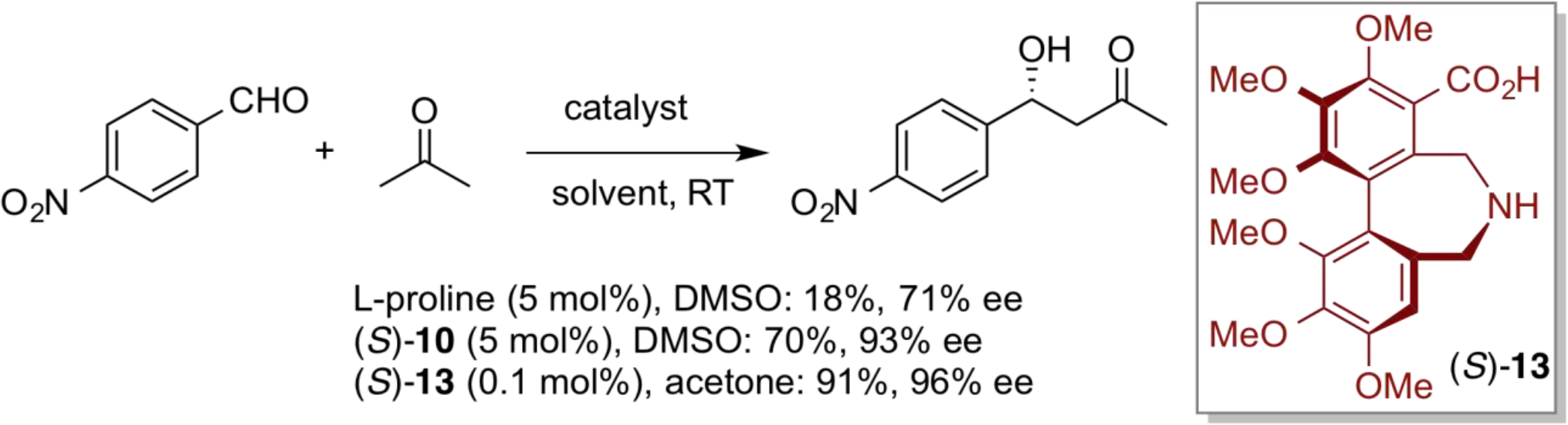
Asymmetric Direct Aldol Reactions
A new artificial amino acid (S)-10 was prepared by introducing a secondary amine moiety and a carboxyl group into a binaphthyl core structure possessing axial asymmetry. The chiral efficiency of newly designed (S)-10 was evaluated by applying a direct asymmetric aldol reaction. Here, proline catalyst showed very low reactivity and moderate enantioselectivity in the direct asymmetric aldol reaction of acetone and p-nitrobenzaldehyde. In marked contrast, however, use of (S)-10 under otherwise similar reaction conditions provided the good yield and high enantioselectivity. Furthermore, optically pure biphenyl-derived catalyst (S)-13 with six electron-donationg methoxy substituents exhibited unusually high reactivity, and with very low catalyst loading (0.1 mol%) high chemical yield and excellent enantioselectivity were observed.
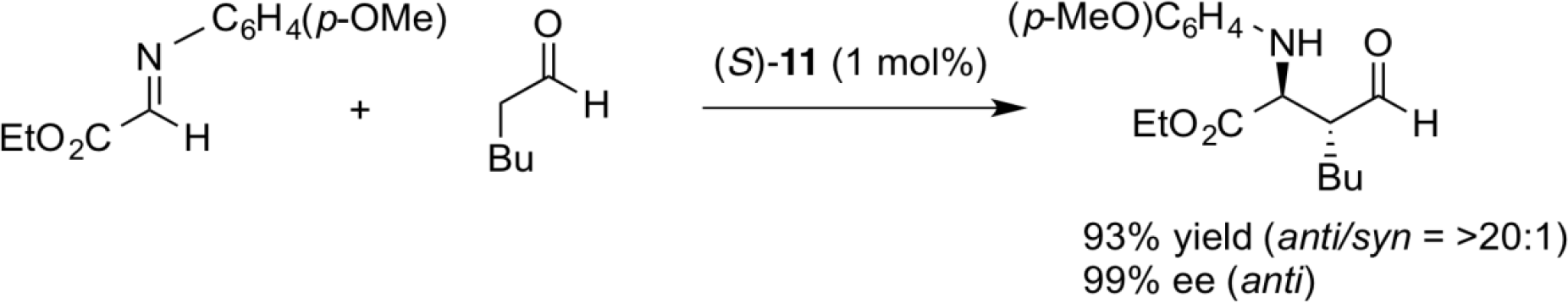
anti-Selective Asymmetric Mannich Reactions
The Mannich reaction is an important carbon-carbon bond forming reaction that gives a β-aminocarbonyl compound. In recent years, many direct asymmetric Mannich-type reactions using various organocatalysts have been reported. When proline is used as organocatalyst, syn-Mannich products are generally obtained with high enantioselectivity via an enamine intermediate with s-trans conformation. In marked contrast, however, our newly designed aminosulfonamide catalyst (S)-11 afforded the opposite anti-Mannich products with high enantioselectivity via an enamine intermediate with s-cis conformation. This reaction proceeds very smoothly even with very low catalyst loading (~0.2 mol%). In addition, the catalyst is stable and can be easily recovered and reused for the next runs. This catalyst is now commercially available from Tokyo Chemical Industry.
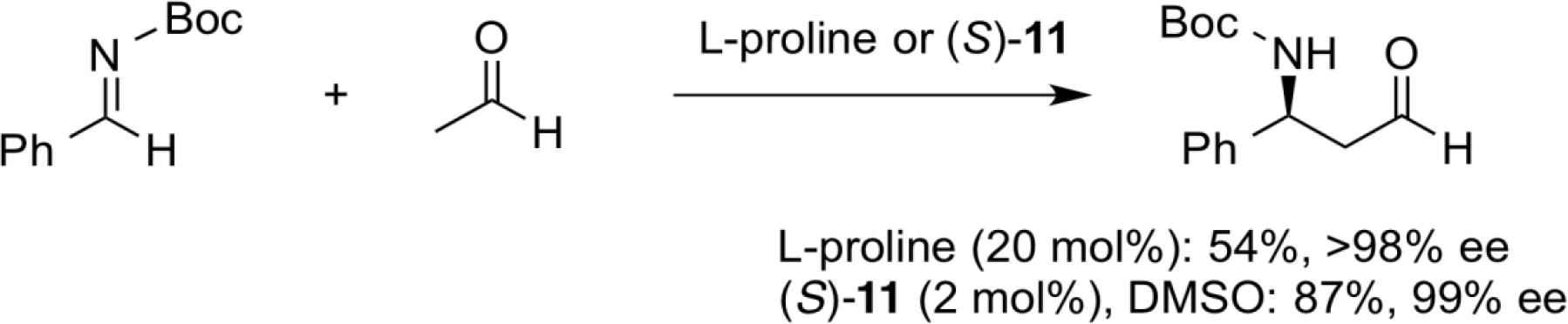
Asymmetric Direct Mannich Reactions
In the direct asymmetric Mannich reaction of highly reactive acetaldehyde, a side reaction such as aldol reaction causes a decrease in the yield of the desired Mannich product. In contrast, our secondary amino sulfonamide catalyst (S)-11 enabled the low catalyst loading with similar excellent enantioselectivity.
Asymmetric Direct Hydroxyamination
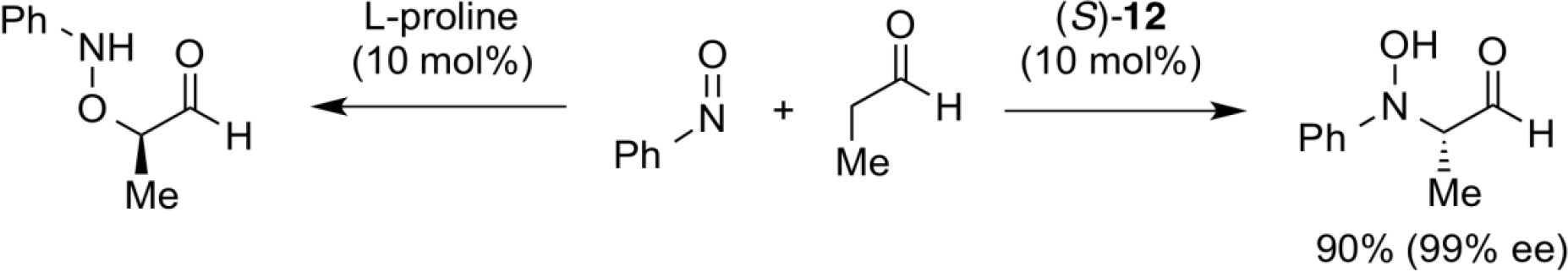
The proline-catalyzed addition of aldehyde to nitroso compound yields an aminohydroxyl compound having a bond formed on the oxygen side of the N=O moiety. On the other hand, the use of chiral binaphthyl-modified secondary amine catalyst (S)-12, in which a bulky tertiary alcohol moiety is introduced into the binaphthyl core structure, showed the opposite regioselectivity on the addition to N=O moiety, resulting in a direct asymmetric hydroxyamination reaction with high enantioselectivity.
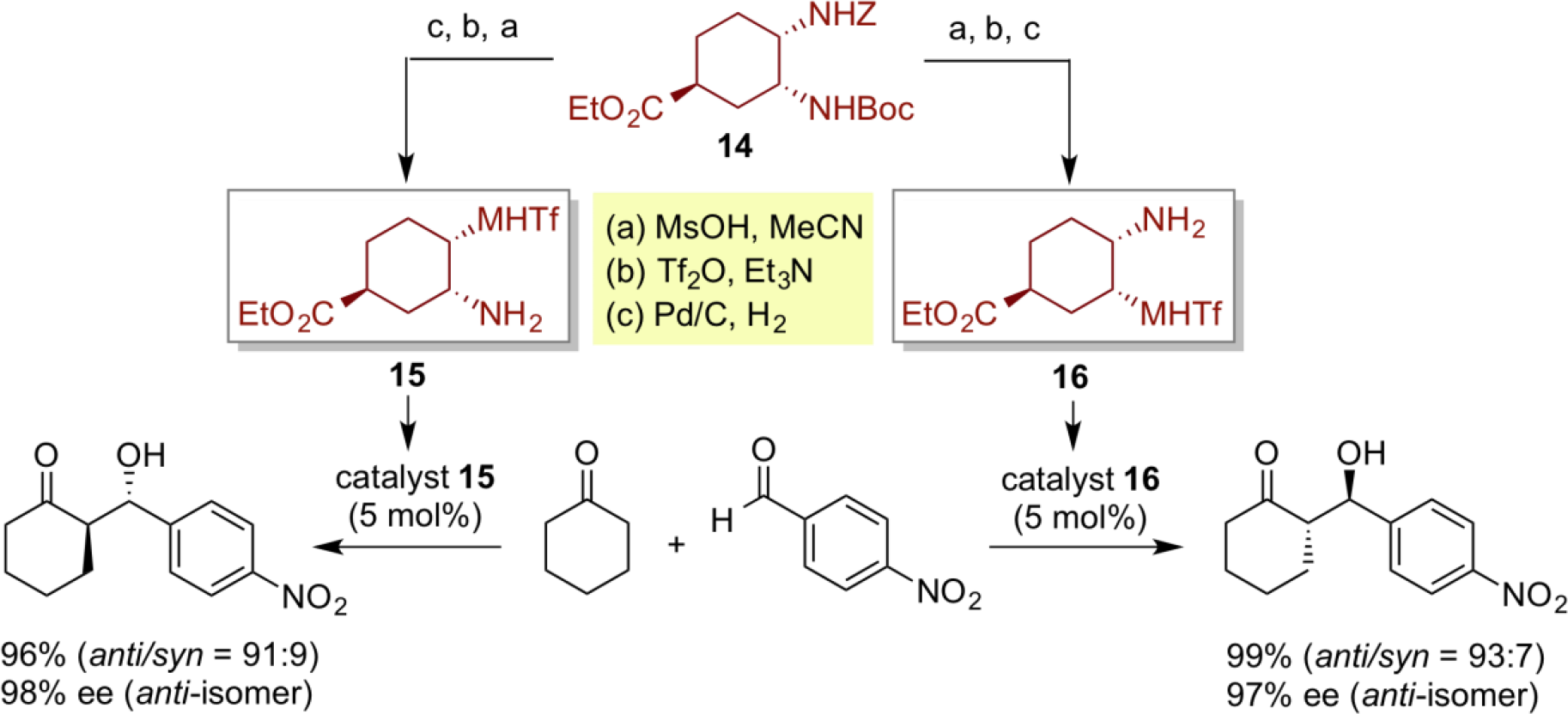
In asymmetric catalysis, both enantiomeric products can be generally prepared from both enantiomeric catalysts, respectively. However, if two different organocatalysts can be prepared from the common chiral sources in a short-step sequnce, and can be successfully applied to the asymmetric synthesis of both enantiomeric products, it would be very practical from the viewpoint of an efficient mass production process of useful chiral materials. Verification of our hypothesis is exemplified by the design of six-membered cis-diamine-type bifunctional organic catalysts 15 and 16. The characteristic feature of such six-membered ring cis-diamine-type bifunctional organocatalyst is that, unlike the conventional organocatalysts, two different chiral difunctional organocatalysts 15 and 16 can be created from the common chiral source 14 in a 3-step sequence. When these two different bifunctional organocatalysts are respectively applied for direct asymmetric aldol reactions, both enantiomeric aldol products are readily obtainable with high enantioselectivity.
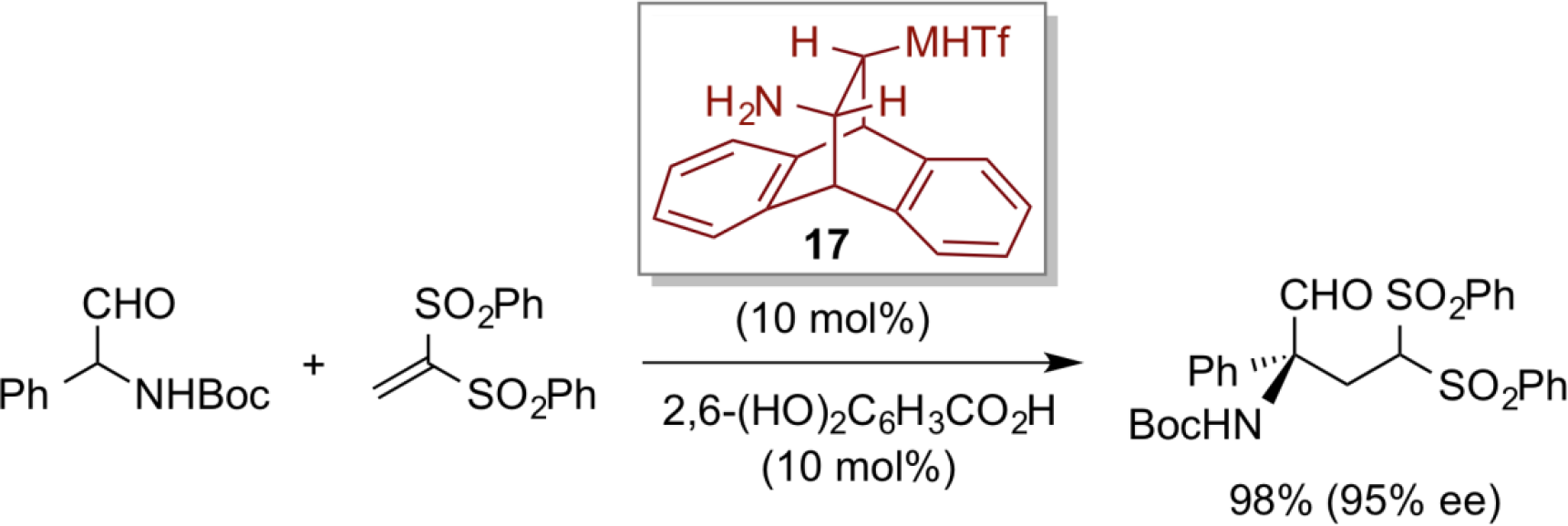
In addition, we have designed a trans-diamine-type bifunctional organic catalyst 17 derived from anthracene, and have successfully utilized for a highly enantioselective conjugated addition of α-aminoaldehyde with vinyl sulfone substrate.
Organoradical Catalysts
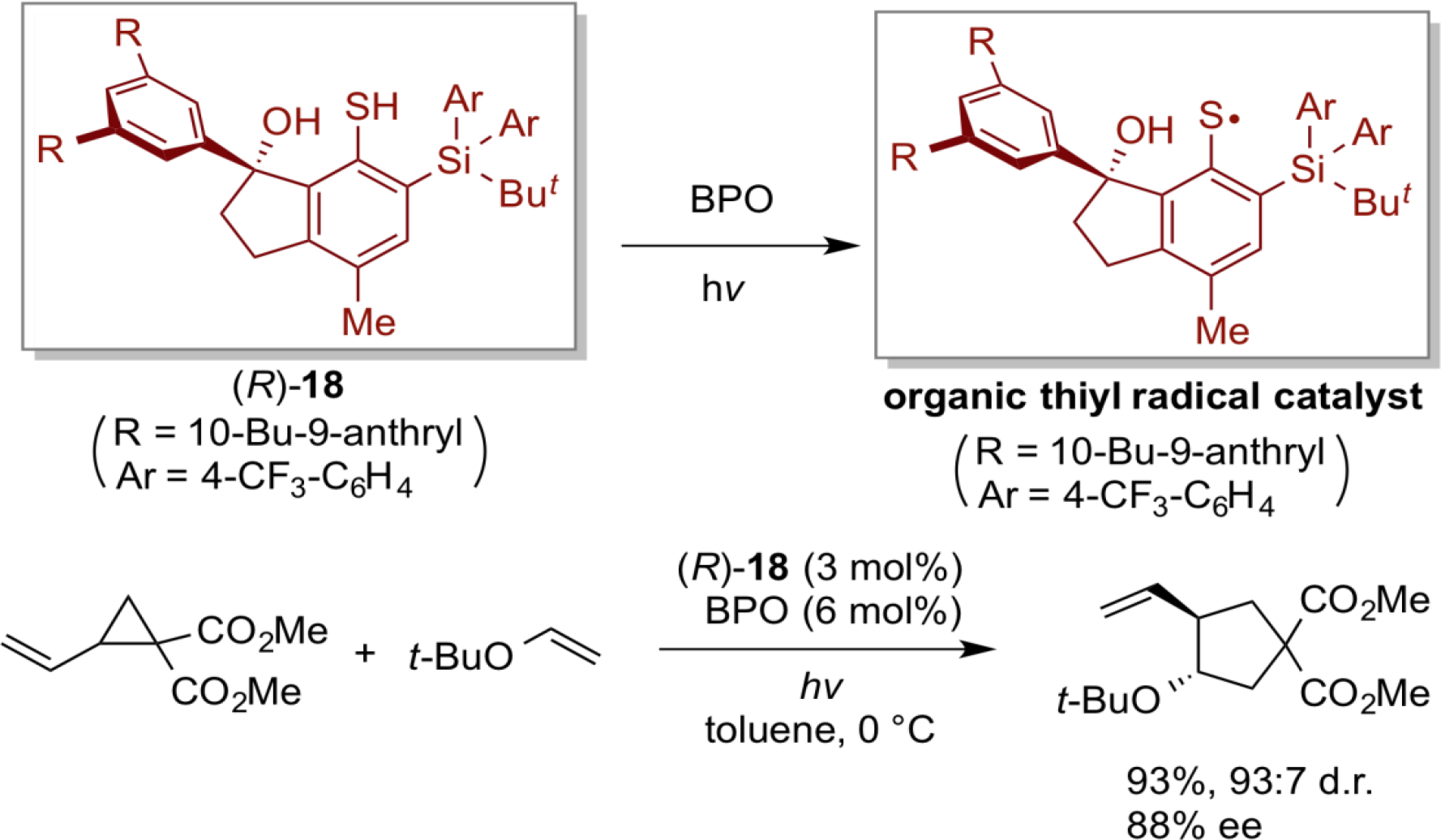
Radicals are highly reactive chemical species, and hence reactions involving radical species are quite attractive, and enable unique transformations that are difficult to achieve with ordinary ionic reactions. However, research on the radical reactions promoted by organoradical catalysts is significantly delayed compared to other organocatalytic fields. In order to develop our own organoradical chemistry, we focused on the new design of organosulfur radicals that can be easily generated from the corresponding disulfides or thiols, and applied such organoradical species to the development of new organoradical reactions in which chiral sulfur radicals behave as active species. As a result, the chiral organosulfur radical generated from chiral thiol (R)-18, having a chiral indanol skeleton, and benzoyl peroxide (BPO) under the irradiation of light effected the asymmetric radical cyclization between vinylcyclopropane and olefin to furnish various chiral polysubstituted cyclopentanes with high yield and high stereoselectivity.
The chiral indanol skeleton, thus created in this study, was found to be an important privileged structure in the design of other organocatalysts. Indeed, organoselenium catalyst 9 and organoiodine catalyst 19 with a similar key skeleton also showed high asymmetric induction in the asymmetric lactonizationof β,γ-unsaturated carboxylic acids and asymmetric dearomatization of phenols, respectively.
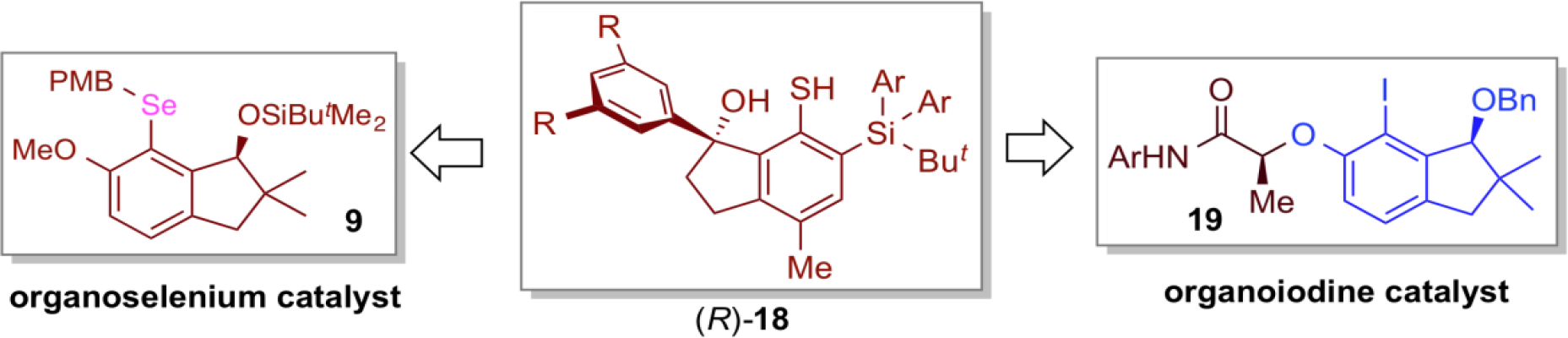
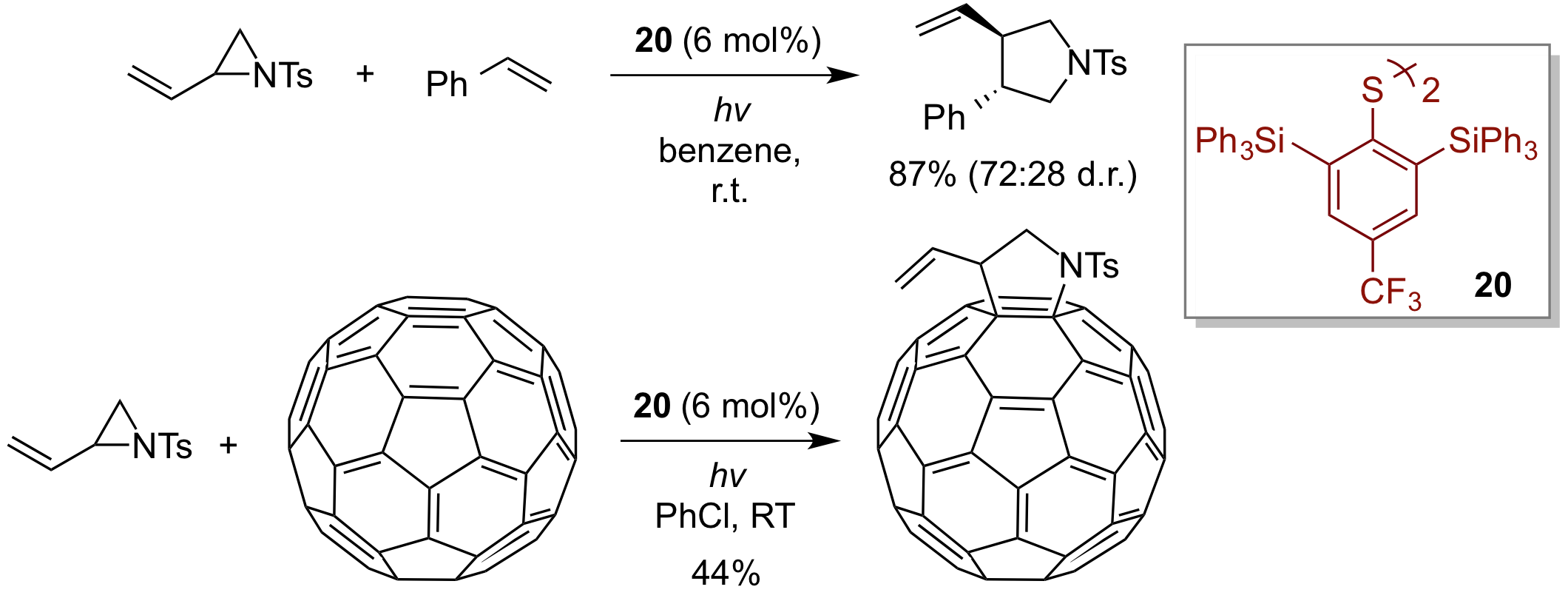
In addition, we have succeeded in developing radical cyclization between vinyl aziridine, which is a nitrogen-containing three-membered ring compound, and olefin by using sterically bulky disulfide 20 as an organosulfur catalyst. This procedure has been found to be useful for the functionalization of fullerenes.
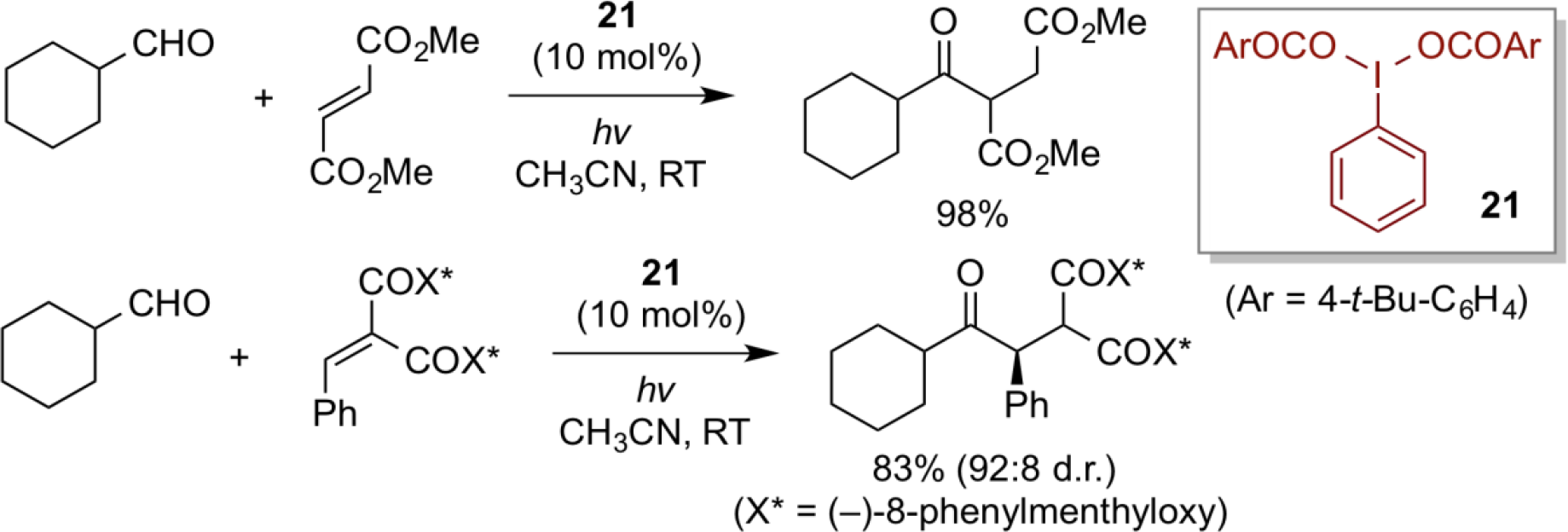
Direct acyl radical formation of linear aldehydes (RCH2-CHO) and subsequent hydroacylation with electron-deficient olefins can be effected with various types of metal and non-metal catalysts/reagents. In marked contrast, however, no successful examples on the use of branched aldehydes have been appeared thus far, because of their strong tendency of generating alkyl radicals through the facile decarbonylation of acyl radicals. Here, use of a hypervalent iodine(III) catalyst of type 21 under visible light photolysis allows a mild way of generating acyl radicals from various branched aldehydes, thereby, by reacting with electron-deficient olefins, giving the corresponding hydroacylated products almost exclusively. Another characteristic feature of this approach is the catalytic use of hypervalent iodine(III) reagent, which is a rare example on the generation of radicals in hypervalent iodine chemistry. In the presence of optically pure unsaturated malonates having (-)-8-phenylmenthyl auxiliary, diastereoselective addition of acyl radical can be realized with high diastereoselectivity.
 pagetop
pagetop