脳内出血治療薬開発を目指した
トロンビン誘発遅延性神経細胞死タンパクの探索
薬品作用解析学分野 藤本 真二
研究指導主任 : 薬品作用解析学分野 教授 赤池 昭紀
研究指導協力者 : 病態機能分析学分野 教授 佐治 英郎
研究内容
研究計画
脳内出血には未だ有効な治療法が確立されておらず、予後は悪く致死率も高い。脳内出血発作時には、脳実質中に漏出した血液由来成分が障害の惹起に寄与していることが報告されている。このうち、血液凝固因子としても知られているトロンビンは、中枢神経系に広く分布しているトロンビン受容体を介して神経細胞およびグリアに作用する。申請者は、生体脳に近い実験系で細胞構築が保持されている培養大脳皮質線条体切片を用いた検討により、トロンビンにより誘発される細胞障害はextracellular signal-regulated kinase (ERK)リン酸化阻害薬およびタンパク合成阻害薬により抑制されることを見いだした。このことから、トロンビン毒性にはERKを介した新規タンパク合成が必須であることが示唆された。
一方、ERKにより誘導される細胞内情報伝達経路は、トロンビンにより誘発される障害だけでなく、細胞の恒常性の維持にも重要な役割を果たしている。それゆえ、トロンビン刺激によるERKリン酸化シグナルの下流において特異的に新規合成されるタンパクの特性を解明することは脳内出血に対する有効かつ安全な治療薬を開発する上で重要な情報となりうる。
この目的において、3H標識したアミノ酸を培養大脳皮質線条体切片に適用した後にトロンビン処置を負荷し、回収した切片を破砕することによって得られる抽出物から、新規合成され3H標識されたアミノ酸を組み込んだタンパクを探索し、その分子量および等電点を電気泳動により導出することには重要な意義がある。トロンビン無処置群との比較によりトロンビンにより合成が亢進するタンパクの物理化学的特性の一部が明らかになると考えられる。また、抽出物の分画操作により放射活性の高い画分を回収し、既知の化学物質との結合実験を行うことによりこのタンパクに対して阻害活性を有する化合物の創製に向けた有用な知見が得られると考えられる。
序論
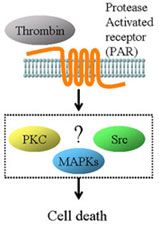 脳内出血発作時には、脳実質中に漏出した血液由来成分が障害の惹起に寄与していることが報告されている。このうち、血液凝固因子としても知られているトロンビン(thrombin)は、血液中に存在するプロトロンビンの切断により合成され、中枢神経系に広く分布しているprotease activated receptor(PAR)を介して神経細胞およびグリアに作用する。培養アストロサイトを用いたこれまでの報告から、トロンビンにより誘発される細胞死においてprotein kinase C(PKC)、チロシンキナーゼ、mitogen-activated protein kinases(MAPKs)が関与していることが示唆されている。また、ミクログリアにおいてトロンビンがMAPKsを介してNO合成酵素(NOS)の発現を誘導し神経細胞を障害する活性酸素種の生成に寄与していることが報告されているが、これらの詳細な機序は明らかになっていない。そこで本研究において、トロンビン誘発細胞障害の機序を解明することが脳内出血に対する治療法を確立する上で重要であると考え、細胞構築が維持されている培養大脳皮質線条体切片を用いた検討を行った。 脳内出血発作時には、脳実質中に漏出した血液由来成分が障害の惹起に寄与していることが報告されている。このうち、血液凝固因子としても知られているトロンビン(thrombin)は、血液中に存在するプロトロンビンの切断により合成され、中枢神経系に広く分布しているprotease activated receptor(PAR)を介して神経細胞およびグリアに作用する。培養アストロサイトを用いたこれまでの報告から、トロンビンにより誘発される細胞死においてprotein kinase C(PKC)、チロシンキナーゼ、mitogen-activated protein kinases(MAPKs)が関与していることが示唆されている。また、ミクログリアにおいてトロンビンがMAPKsを介してNO合成酵素(NOS)の発現を誘導し神経細胞を障害する活性酸素種の生成に寄与していることが報告されているが、これらの詳細な機序は明らかになっていない。そこで本研究において、トロンビン誘発細胞障害の機序を解明することが脳内出血に対する治療法を確立する上で重要であると考え、細胞構築が維持されている培養大脳皮質線条体切片を用いた検討を行った。
実験方法
- 培養大脳皮質線条体切片の作製
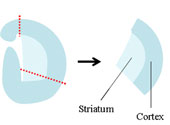 3〜4日齢のWistar系ラット新生仔の全脳を摘出し、厚さ300 µmの大脳皮質線条体冠状断切片を作製し、34゚C、5%CO2環境下、多孔質膜上で静置界面培養した。培地の組成は、50% Minimum Essential Medium、25% Hanks’ Balanced Salt Solution、25% ウマ血清とした。 3〜4日齢のWistar系ラット新生仔の全脳を摘出し、厚さ300 µmの大脳皮質線条体冠状断切片を作製し、34゚C、5%CO2環境下、多孔質膜上で静置界面培養した。培地の組成は、50% Minimum Essential Medium、25% Hanks’ Balanced Salt Solution、25% ウマ血清とした。
- 薬物処置
培養9〜11日目に培地を無血清培地に交換し、その24〜48時間後にthrombin、薬物およびpropidium iodide (PI; 5 µg/ml)を含有した無血清培地で72時間培養した。一部の実験においてはthrombin曝露の24時間前から薬物を投与した。
- 細胞障害の評価
薬物処置開始後24、48、72時間後の大脳皮質内単位面積あたり(180 µm×180 µm)のPIの蛍光強度を測定し、NMDA (100 µM)を72時間投与したときの蛍光強度を100%として、相対的な蛍光強度を算出し、評価した。一部の実験においては切片全体の画像を取得し、線条体の面積を測定した。
- 免疫組織化学
薬物処置3時間後あるいは72時間後に切片を4% paraformaldehydeで固定し、一次抗体として各細胞マーカーおよびphosphorylated extracellular signal-regulated kinase (pERK)に対する抗体を用い、蛍光標識した二次抗体を反応させた後、共焦点顕微鏡により観察した。一部の実験では二次抗体と同時にHoechst33342 (0.1 mg/ml)を添加した。
- Western Blotting
薬物処置した切片をlysis bufferにより回収した後、タンパク量を一定にしてSDS化した。SDS-PAGEによりタンパクを分離し、PVDF膜に転写した。各抗体と反応させた後、ECLによりタンパクを検出した。
結果
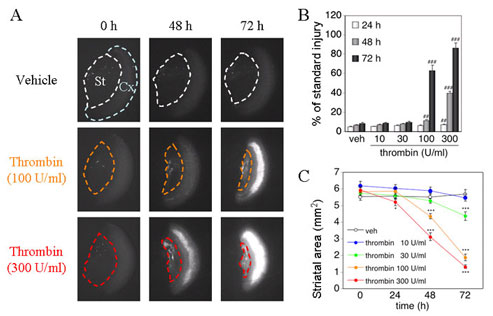
Fig.1 Thrombin-induced cortical cell injury and striatal shrinkage. Scale bar, 1 mm. ## P < 0.01,
### P < 0.001 vs. vehicle (veh) at respective time points. * P < 0.05, *** P < 0.001 vs. 0 h of respective treatment.
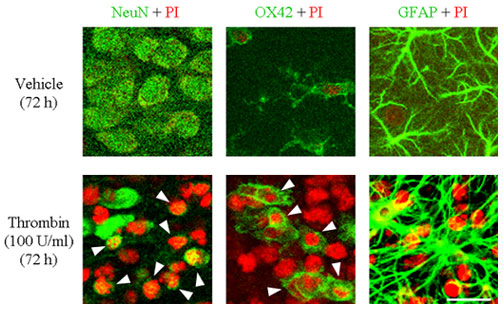
Fig.2 Confocal images of the cortical region of slice cultures exposed to vehicle (upper) and thrombin (100 U/ml) (lower) for 72 h and immunostained for cell type-specific markers. Scale bar, 20 µm.
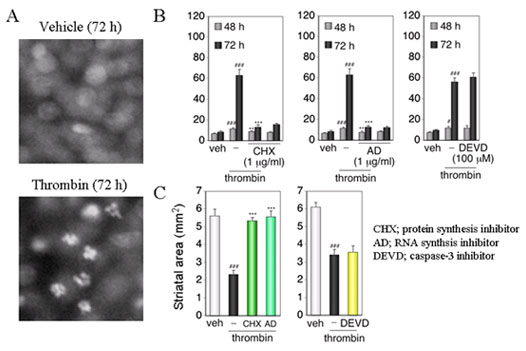
Fig.3 Thrombin-induced cell injury accompanies nuclear fragmentation, and is dependent on new protein synthesis but not on caspase activity. # P < 0.05, ### P < 0.001 vs. vehicle, * P < 0.05,
** P < 0.01, *** P < 0.001 vs. thrombin alone.
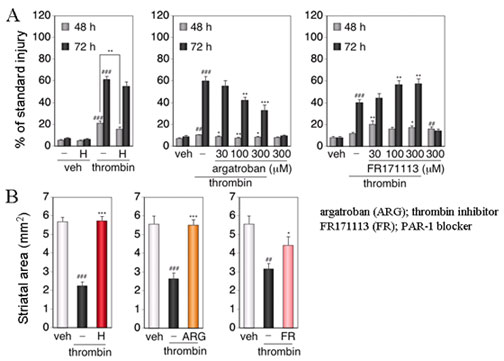
Fig.4 Involvement of protease activity and protease activated receptor (PAR)-1 activation in thrombin cytotoxicity. ## P < 0.01, ### P < 0.001 vs. vehicle, * P < 0.05, ** P < 0.01, *** P < 0.001 vs. thrombin alone.
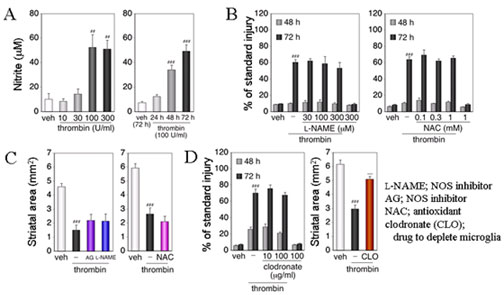
Fig.5 Involvement of nitric oxide production, oxidative stress and microglia activation in thrombin toxicity. ## P < 0.01, ### P < 0.001 vs. vehicle, *** P < 0.001 vs. thrombin alone.
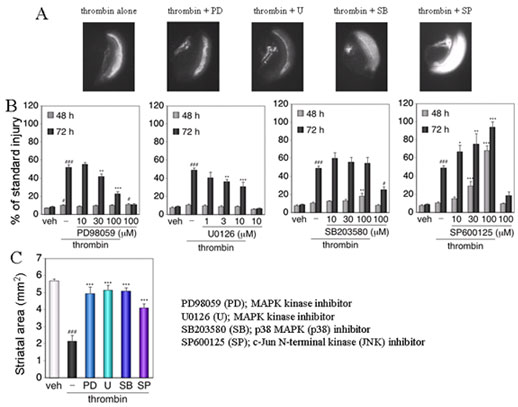
Fig.6 Differential involvement of MAPKs in thrombin toxicity in the cortex and in the striatum.
# P < 0.05, ### P < 0.001 vs. vehicle, * P < 0.05, ** P < 0.01, *** P < 0.001 vs. thrombin alone.
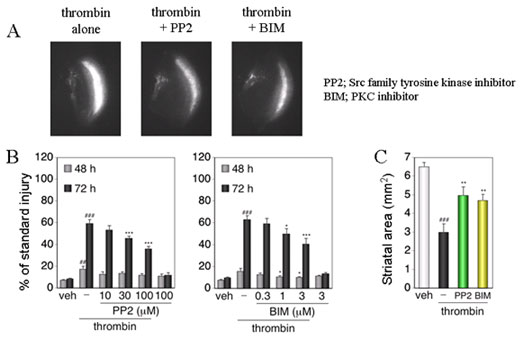
Fig.7 Effects of inhibitors of Src and PKC on thrombin cytotoxicity. ## P < 0.01, ### P < 0.001 vs. vehicle, * P < 0.05, ** P < 0.01, *** P < 0.001 vs. thrombin alone.
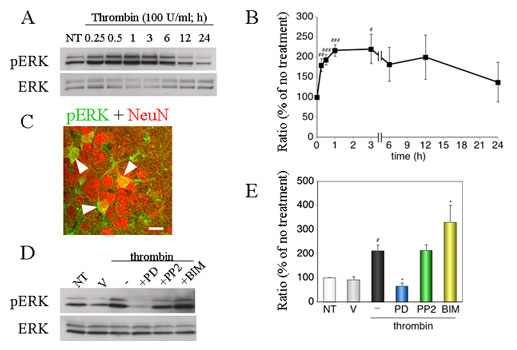
Fig.8 Thrombin-induced phosphorylation of ERKs. Scale bar, 20 µm.
# P < 0.05, ## P < 0.01, ### P < 0.001 vs. time 0, * P < 0.05 vs. thrombin alone.
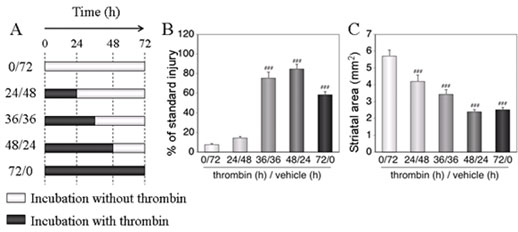
Fig.9 Persistent signaling is required for induction of cell death by thrombin. ### P < 0.001 vs. vehicle (indicated as “0/72”).
総括および今後の展望
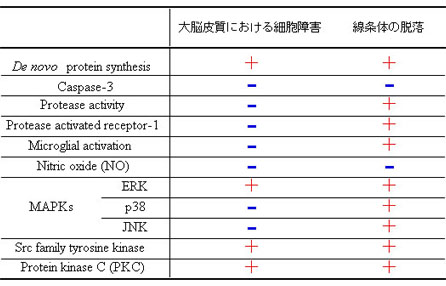
- SrcおよびPKCの阻害はERKのリン酸化を抑制しなかった。
- トロンビンにより誘発される障害にはERKのリン酸化のタイムコースよりも長い刺激が必要であった。
以上の結果から、トロンビンにより誘発される障害には、ERK、Src型チロシンキナーゼおよびPKCを介した新規タンパク合成が重要な役割を担っていることが示唆された。ERKのリン酸化はSrcファミリーチロシンキナーゼおよびPKCの活性化を介しておらず、プロテアーゼ活性に依存しない経路を含めた複数の経路により障害が誘発されることが考えられる。また、トロンビンは大脳皮質および線条体において異なった形態の障害を惹起し、この障害は一部の薬物に対して異なった応答をしたことからトロンビンにより誘発される障害は脳の部位によって異なった機序を介していることが示唆された。今後、トロンビンにより誘発される障害の機序をさらに詳細に検討し、リン酸化ERKの下流のシグナルとして新規に合成されるタンパクを同定することとともに、in vivo において脳内出血好発部位においてトロンビンを始めとするセリンプロテアーゼが脳組織に及ぼす影響を検討することは脳内出血治療薬を開発する上で重要であると考えられる。
|
