Research
[2018] [2017] [2016] [2015] [2014] [2013] [2012] [2011] [2010] [2009] [2008] [2007] [2006]
2008
Total synthesis of (±)-lysergic acid, lysergol, and isolysergol by palladium-catalyzed domino cyclization of amino allenes bearing a bromoindolyl group.
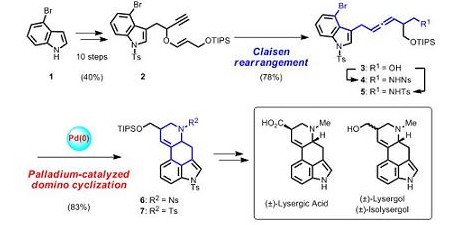
Ergot alkaloid類はカビの菌核より抽出されたインドールアルカロイドであり、リゼルグ酸をはじめとして数多くの天然物が報告されています。今回我々は、パラジウム触媒によるアレンの連続環化反応を鍵反応
としたergot alkaloid骨格の合成を計画しました。市販の4-bromoindoleから10工程で合成したpropargyl vinyl ether 2に対して金触媒によるClaisen転位反応を行うことでアレン3を合成しました。3をノシルアミド4お
よびトシルアミド5に変換後、パラジウム触媒を用いた連続環化反応により、ergot alkaloid骨格を有する6,7を良好な収率で得ることに成功しました。さらに官能基変換により生理活性天然物であるlysergol、isolyserg
olおよびlysergic acid,の全合成を達成しました。[PubMed]
Org. Lett.10(22) 5239-5242 (2008)
Development of novel GPR54 agonists with resistance to degradation by matrix metalloproteinase.
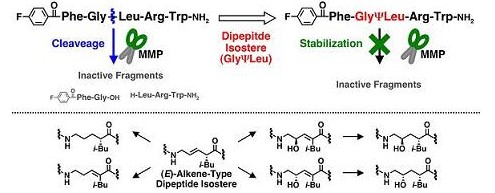
-マトリックスメタロプロテアーゼ耐性新規GPR54アゴニストの開発-
当分野において見出されたGPR54アゴニストは強力な生理活性を有するものの、マトリックスメタロプロテアーゼ(MMP)によりGly-Leuペプチド結合が切断され、失活してしまいます。そこで、この加水分解部
位をアミド等価体に変換したMMP耐性GPR54アゴニストを設計しました。GPR54アゴニスト活性を維持することのできる非水解性構造を効率的に見出すため、E型アルケンジペプチドイソスターを共通中間体
として種々のジペプチドイソスターを合成し、ペプチド鎖に導入しました。その結果、GPR54アゴニスト活性を損なうことなく生体内安定性が改善された新規GPR54アゴニ
ストを見出しました。[PubMed]
J. Med. Chem.51(23) 7645-7649 (2008)
Identification of minimal sequence for HIV-1 fusion inhibitors.
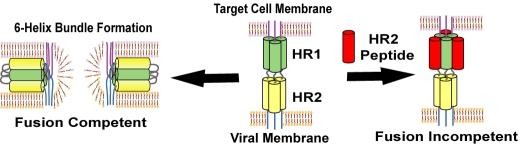
エイズ治療において既存の薬剤に対する耐性ウイルスの出現が問題となっており、新規標的としてウイルス-宿主膜融合過程が注目されています。HIVウイルス由来ペプチドのC34, T-20はヘリックス性が高く、抗HIV活
性を示すことが知られています。本研究では抗HIV活性に必要な最少構造を探索し、C34のC末端5残基を除きへリックスを誘起するモチーフを導入したSC29EKが高い抗ウイルス活性を維持することを見出しました。さら
にヘリックス構造の安定化及びプロテアーゼ耐性を目的として溶媒接触面のアミノ酸残基を非天然アミノ酸に変換し、活性を維持した誘導体のデ
ザインに成功しました。[PubMed]
Bioorg. Med. Chem. 16(20) 9184-9187 (2008)
Concise synthesis of indole-fused 1,4-diazepines through copper(I)-catalyzed domino three-component coupling-cyclization-N-arylation under microwave irradiation.
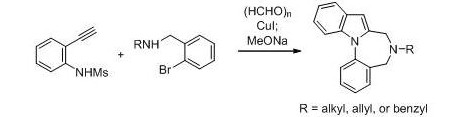
現代の有機合成化学は、入手容易な化合物から複雑な構造を有する骨格を一挙に構築する方法論の開発が求められています。特に、一つのフラスコ内で複数の素反応を触媒するタンデム触媒は、環境負荷が小
さく原子効率に優れた反応として注目されています。今回我々は、有用な創薬テンプレートとして知られているインドール縮環型1,4*ベンゾジアゼピン骨格のタンデム触媒型一挙構築法の開発を行いました。検討の結果、
入手容易なヨウ化銅をタンデム触媒として、2-エチニルアニリン誘導体、パラホルムアルデヒド、o-ブロモベンジルアミンの三成分反応*インドール形成*N-アリール化により、インドール縮環型1,4*ベンゾジアゼピンを
一挙に合成することに成功しました。
Org. Lett. 10(16) 3535-3538 (2008)
Identification of novel nonpeptide CXCR4 antagonists by ligand-based design approach.
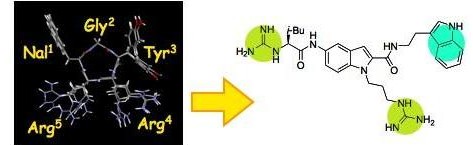
一般に、ペプチド性化合物は、経口吸収性や体内動態特性がすぐれない ことから、そのすぐれた生物活性にも関わらず、創薬展開が敬遠される 傾向にあります。我々は、ケモカイン受容体CXCR4に対し
て強力 なアンタゴニスト活性を示す環状ペンタペプチドFC131につい て、小分子化合物への変換を試み、ドラッグ-ライクテンプレー ト上にFC131のファルマコフォアの空間提示の再現を期待した 様々な誘導
体の中から、中程度のCXCR4アンタゴニスト活性を示 す非ペプチド性化合物を見出しました。
Bioorg. Med. Chem. Lett. 18(14) 4124-4129 (2008)
Palladium-catalysed biscyclisation of allenic bromoalkenes through zipper-mode cascade.
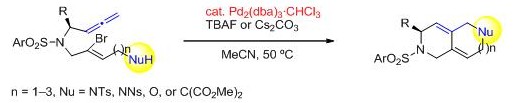
アレンとアルケニルハライドはパラジウム触媒存在下においてカルボパラデーションによりπ-アリル錯体を形成し、さまざまな求核剤と反応することが広く知られています。しかしながら、分子内に求核部位を有するアル
ケニルハライドを用いた環化反応の例はごく限られており、分子内連続環化反応による縮環型複素環構築に成功した例はこれまで報告されていません。我々は、分子内に求核基を導入したブロモエンアレンに触媒
量のトリス(ジベンジリデンアセトン)ジパラジウムとTBAFをアセトニトリル溶媒中で作用させると縮環型二環性複素環を収率良く得られることを見出しました。さらに本反応は縮環型中員環やベンゾアゼピン骨格を有する
三環性複素環の合成にも広く適用できることが明らかとなりました。
Chem. Commum. 303534-3536 (2008)
Palladium-catalyzed sp3 C*H activation of simple alkyl groups: direct preparation of indoline derivatives from N-alkyl-2-bromoanilines.
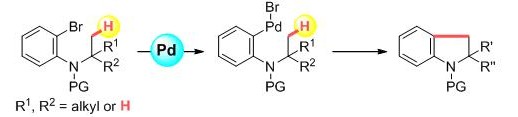
遷移金属触媒を用いたC-H活性化反応は近年盛んに研究が行われていますが、そのほとんどがsp2炭素を有する芳香環のC-H活性化に留まっており、sp3炭素のC-H活性化の例はほとん
ど知られていません。また、これまで報告されてきたsp3C-H活性化反応は、ベンジル位など高活性なC-H結合の反応例を除いて、分子内に配向基や4級炭素を必要とするため基質一般性に乏しいも
のでした。今回、我々はパラジウム触媒を用いて通常上活性であるアルキル基のsp3C-H結合を活性化し新たにC-C結合を形成することでインドリン骨格を構築する新規合成法を開発しました。本反
応は芳香環部位にヘテロ環を導入しても効率的に進行し、分子内に4級炭素を必要とせずにC-H活性化を起こすことができる点において極めて有用です。[Link]
Org. Lett. 10(9) 1759-1762 (2008)
Structure-activity relationship of pyrazine-based CK2 inhibitors: synthesis and biological evaluation of 2,6-disubstituted pyrazine and 4,6-disubstituted pyrimidine derivatives.
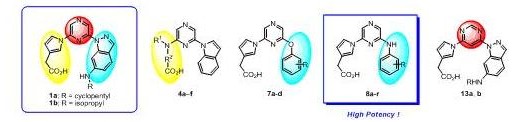
最近、我々はプロテインキナーゼCK2の阻害剤が腎炎の治療薬として有望であることを見出しました。この研究成果に基づいて東レの研究グループによって開発されたCK2特異的阻害剤 (1a, 1b) は母核のピラジン環に
ピロール酢酸部位と6-アミノインダゾール部位が結合した構造を有しています。今回我々は類似の基本構造を有する2,6-二置換ピラジン及び4,6-二置換ピリミジン誘導体を各種合成し、そのCK2阻害活性を評価しました。その結
果、ピロール酢酸と一置換アニリンが結合したピラジン誘導体が高い阻害活性を有することを見出し、構造活性相関についての有用な知見を得ました。
Archiv. Pharm. (Weinheim) 341(9) 554-561 (2008)
Facile synthesis of 3-(aminomethyl)isoquinolines by copper-catalysed domino four-component coupling and cyclisation.
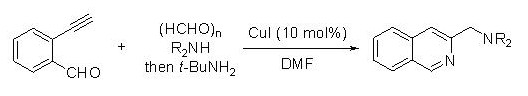
Isoquinoline骨格は生物活性化合物や天然物に広く見られる基本構造です。特に3位にアミノメチル基を有するisoquinolineは重要な生物活性をもつ化合物に存在します。一方で、多成分反応や連続結合形成反応
は、環境調和型反応として注目を集めています。これらの観点から、今回我々は多成分カップリングによる3-(aminomethyl)isoquinolineの合成を検討しました。触媒量のCuI存在下2-ethynylbenzaldehyde、(HCHO)n、二級
アミン、t-BuNH2の四成分を反応させることにより、目的の3-(aminomethyl)isoquinolineを高収率で得ることに成功しました。本反応によって、三つのC-N結合と一つのC-C結合を一挙に構
築できます。[Link]
Chem. Commun. 7835-837(2008)
Diastereoselective synthesis of highly functionalized fluoroalkene dipeptide isosteres and its application to Fmoc-based solid phase synthesis of a cyclic pentapeptide mimetic.
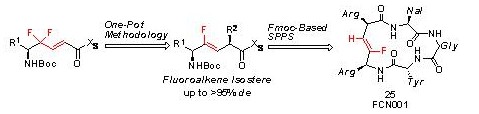
トランスメタル化を経由するOne-Pot還元/上斉アルキル化反応を鍵反応として官能基を有するフルオロアルケン型イソスターの合成およびペプチド固相合成法への応用を検証しました。また、ケモカイン受容
体CXCR4アンタゴニストである環状ペンタペプチド FC131 [cyclo(-D-Tyr-Arg-Arg-Nal-Gly-)]のArg-Arg間のペプチド結合にフルオロアルケン型イソスターを導入し、297 nMと強力なアンタゴニスト活性を有する環状
シュードペプチドFCN001の創出に成功しました。
Tetrahedron, 64(19) 4332-4346 (2008)
Efficient synthesis of trifluoromethyl and related trisubstituted alkene dipeptide isosteres by palladium-catalyzed carbonylation of amino acid-derived allylic carbonates.
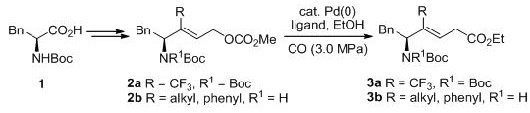
ペプチドリード創薬において、ペプチド結合の非水解性結合への置換は重要な方法論のひとつです。CF3アルケン型ジペプチドイソスター(CF3-ADIs)はペプチド結合に近似した双極子モーメントを有する
イソスターであり、その立体選択的合成法の開発が求められています。今回我々は、フェニルアラニン誘導体1より得られたカルボナート2aに対しパラジウム触媒を用いたカルボニル化反応を行い、目的とす
るPhe-Gly型CF3-ADI 3aを立体選択的に合成することに成功しました。また本合成法を応用し、種々の三置換アルケンジペプチドイソスター3bの合成にも成功しました。[Link]
J. Org. Chem. 73(10) 3942-3945 (2008)
Direct construction of bicyclic heterocycles by palladium-catalyzed domino cyclization of propargyl bromides.
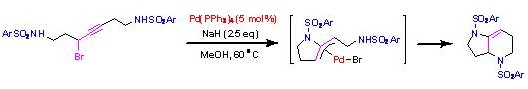
パラジウム触媒により二つの求核種をプロパルギル化合物に導入する反応は広く知られていますが、分子内に二つの求核種を有する基質を用いた二環性複素環の一挙構築に成功した例は報告されていません
でした。我々は、分子の両末端にスルホンアミド求核部位を有するプロパルギルブロミドに対し、触媒量のテトラキス(トリフェニルホスフィン)パラジウムと水素化ナトリウムをメタノール溶媒中で作用させると、縮環型二環性複
素環が収率良く得られることを見出しました。興味深いことに、反応の位置選択性はプロパルギルブロミド部位との位置関係に関係なく、求核種の反応性によって規定されることも明らかになりました
Org. Lett. 10(6) 1171-1174 (2008)
Structure-activity relationship study and NMR analysis of fluorobenzoyl pentapeptide GPR54 agonists.
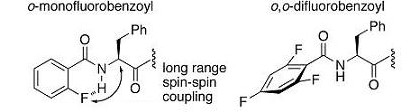
-フルオロベンゾイル基を有するペンタペプチド型GPR54アゴニストの構造活性相関研究とNMR解析-
本研究では強力なGPR54アゴニストとして4-フルオロベンゾイル誘導体の構造展開として、様々なフルオロベンゾイル基を有するペンタペプチドの構造活性相関研究を行いました。その結果、オルト位、メタ位へのフッ素置換は受
容体結合親和性に上利に働くことが明らかとなりました。また、各誘導体のNMR解析より、オルト位にフッ素をひとつ有するベンゾイル基は分子内水素結合を形成することで芳香環とカルボニル基が同一平面上にある一方で、
オルト位にフッ素をふたつ有するものは立体もしくは静電的反発により芳香環平面とカルボニル平面が直交していることが示唆されました。[Link]
Biopolymers, 90(4) 503-511 (2008)
Synthesis and application of fluorescein- and botin-labeled molecular probes for chemokine receptor CXCR4.
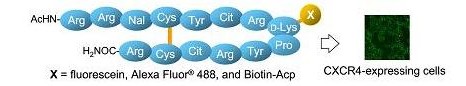
我々はカブトガニ血漿蛋白由来の防御ペプチドpolypphemusin IIをリード化合物として、14残基のβシート様ペプチドT140を開発し、T140がCXCR4に対して強力なアンタゴニスト活性を持つ事を見出しています。こ
れまでの構造-活性相関情報により、T140はArg2、Nal3 (3-(2-naphthyl)alanine)、Tyr5、Arg14の4つの残基がファルマコフォアであることがわかっています。そこでCXCR4への結合能の低下を避けるために、ファルマコフォ
ア外であるN末端側あるいはβターン上のD-Lys8のアミノ基を蛍光標識し、種々のT140蛍光誘導体を合成しました。その結果、D-Lys8のアミノ基を蛍光標識した誘導体において、CXCR4への結合活性の維持が認められ
ました。またこれら得られた誘導体をフローサイトメトリーおよび共焦点顕微鏡を用いてin vitro系内におけるCXCR4への結合確認実験を行ったところ、CXCR4特異的な蛍光イメージングが可能であることが確
認できました。[Link]
ChemBioChem. 9(7) 1154-1158 (2008)
Design of a novel HIV-1 fusion inhibitor that displays a minimal interface for binding affinity.
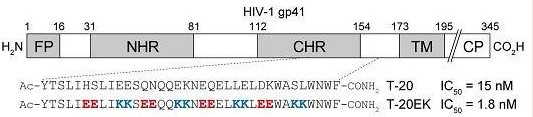
HIV-1はエンベロープタンパクのgp41を介して標的細胞内に侵入します。その過程で、gp41のαへリックス性の高いNHR領域とCHR領域が6- helical bundleを形成することが重要であると知られています。CHR由来ペプチドの
一つであるT-20は、膜融合を阻害することにより抗HIV活性を示すと考えられていますが、詳細なメカニズムは明らかにはなっていません。我々は、分子内での塩橋によりαへリックスを誘起するX-EE-XX-KKの配列をT-20に導入し
た新規阻害剤T-20EKをデザインし、生物活性および物理化学的特性の評価を行いました。T-20EKはヘリックス性の向上に伴い抗HIV活性が向上し、野生型だけでなくT-20耐性ウイルスやHIV-2に対しても高い活性
を示しました。
J. Med. Chem. 51(3) 388-391 (2008)